
CovidShiny: An Integrated Web Tool for SARS-CoV-2 Mutation Profiling and Molecular Diagnosis Assay Evaluation In Silico

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
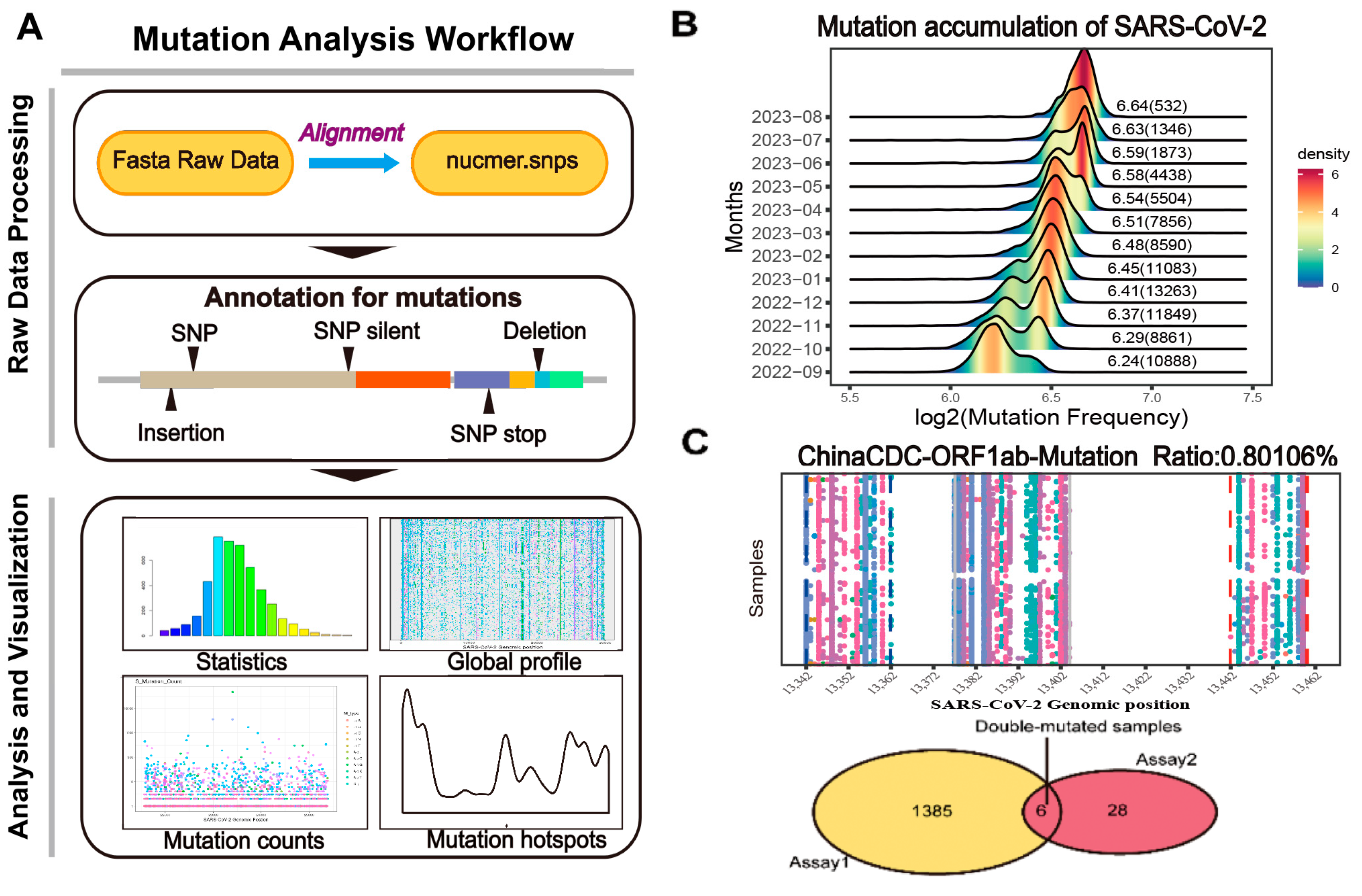
2.1. Data Collection, Pre-Process, and Curation
2.2. Interactive Modules
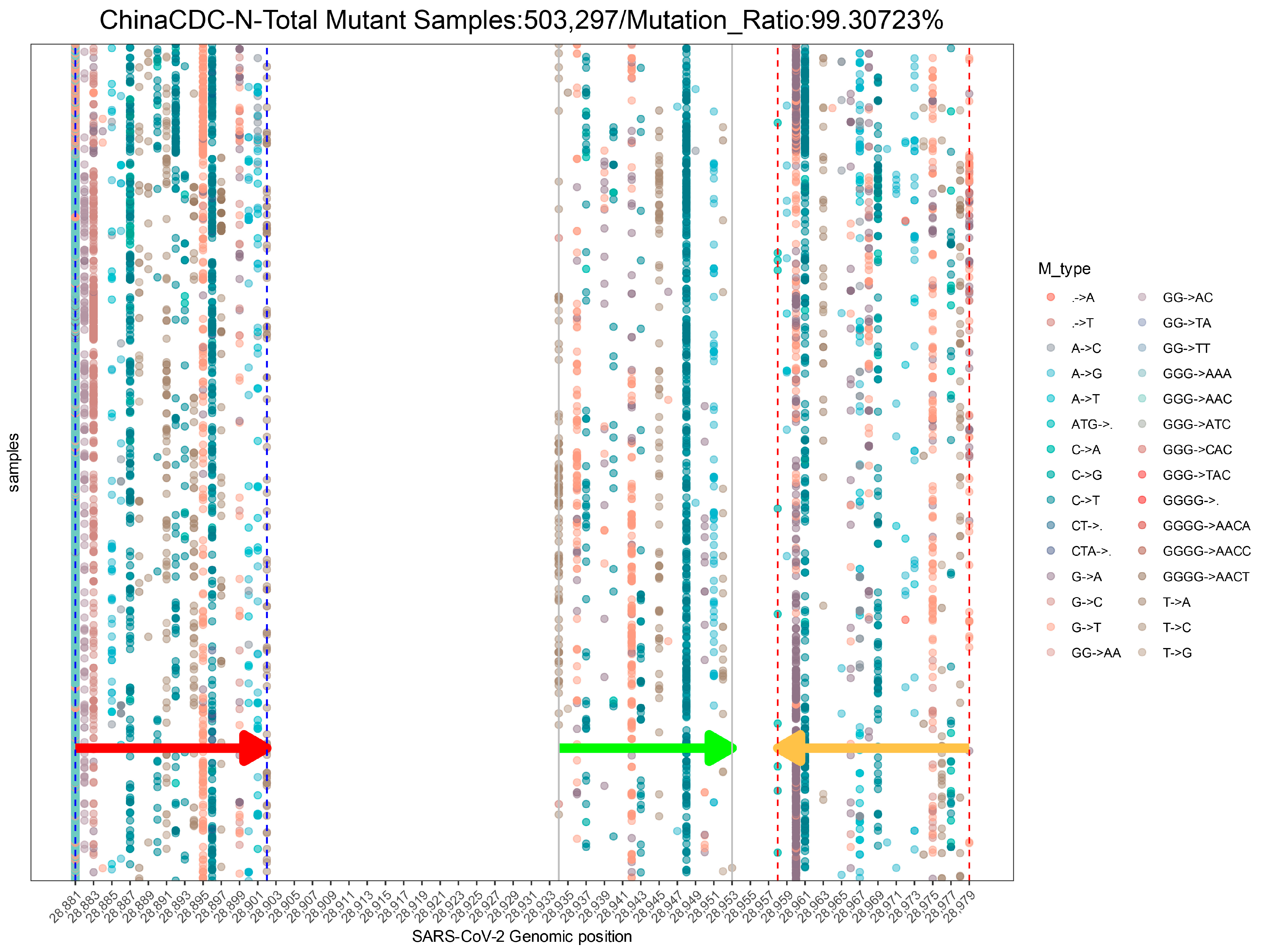
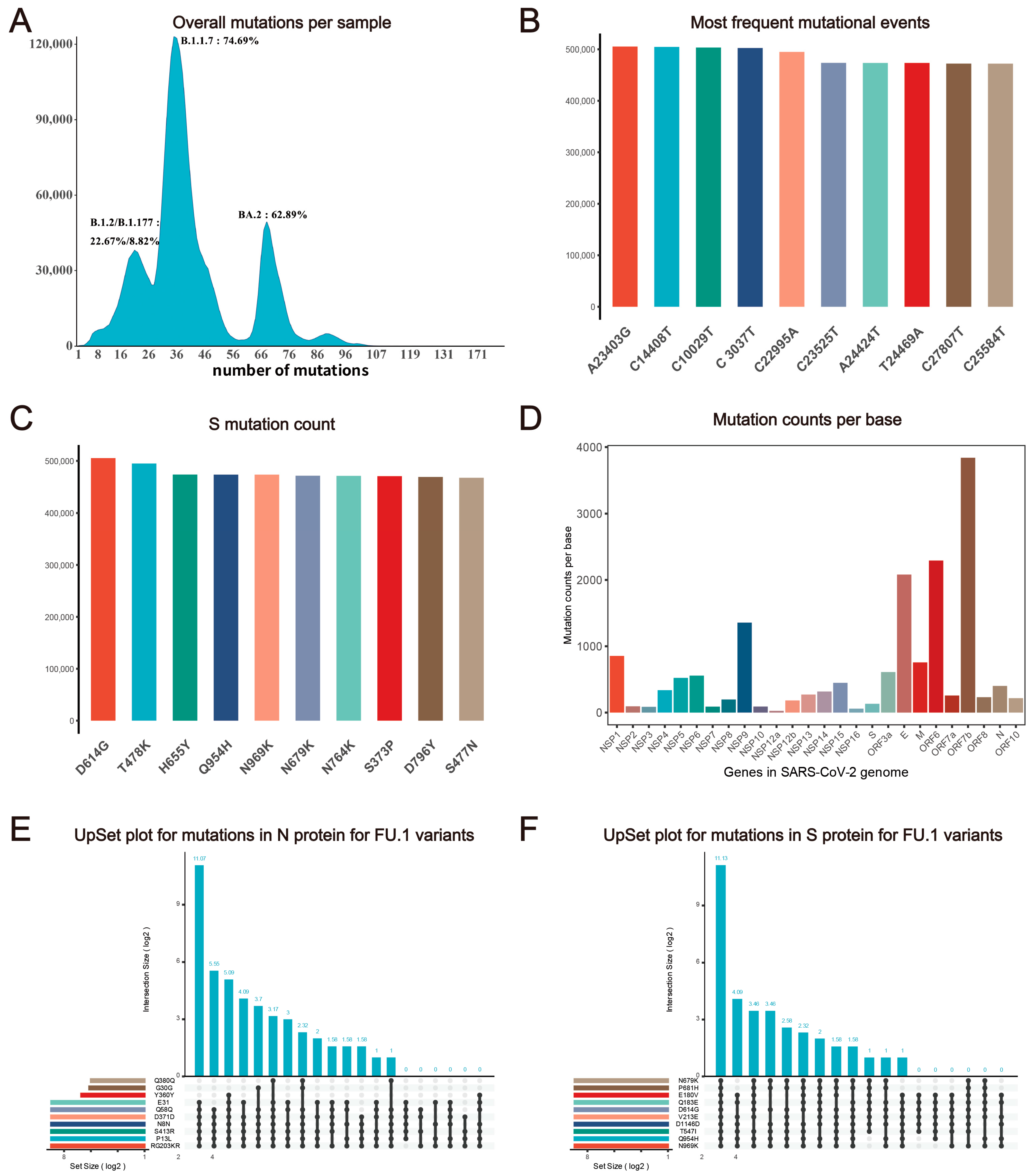
2.2.1. Mutation Statistics
2.2.2. SNP and Protein Profile
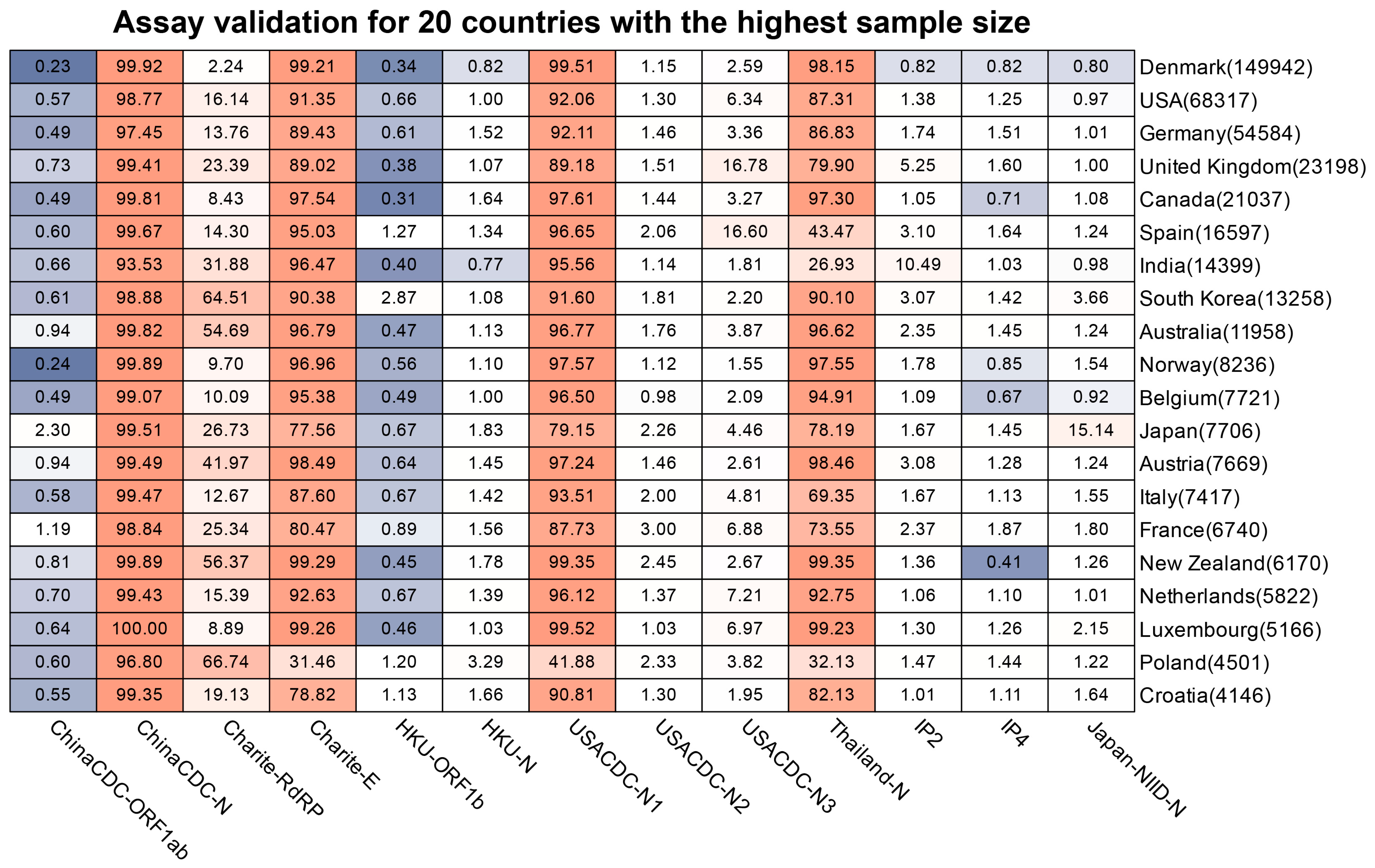
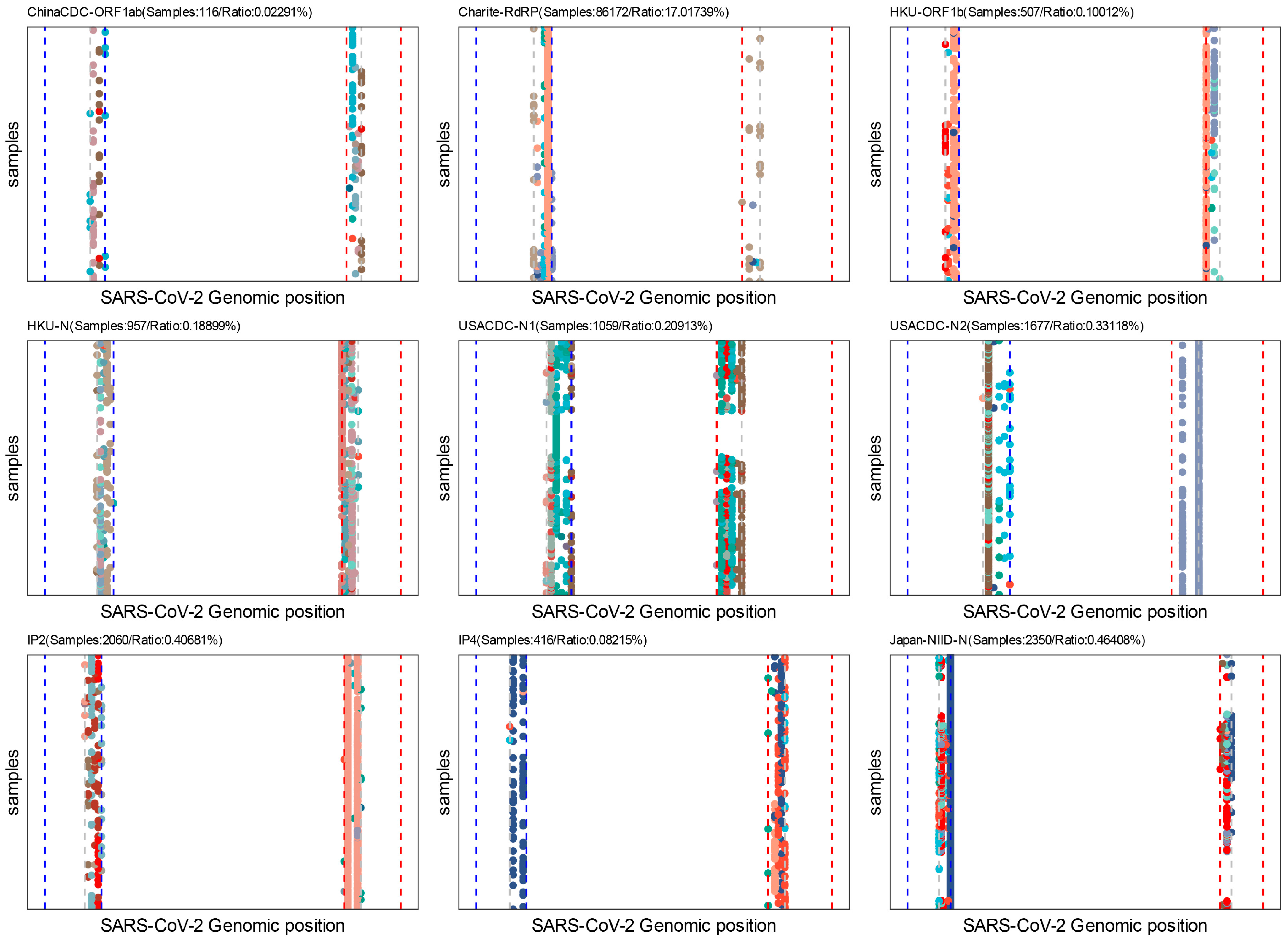
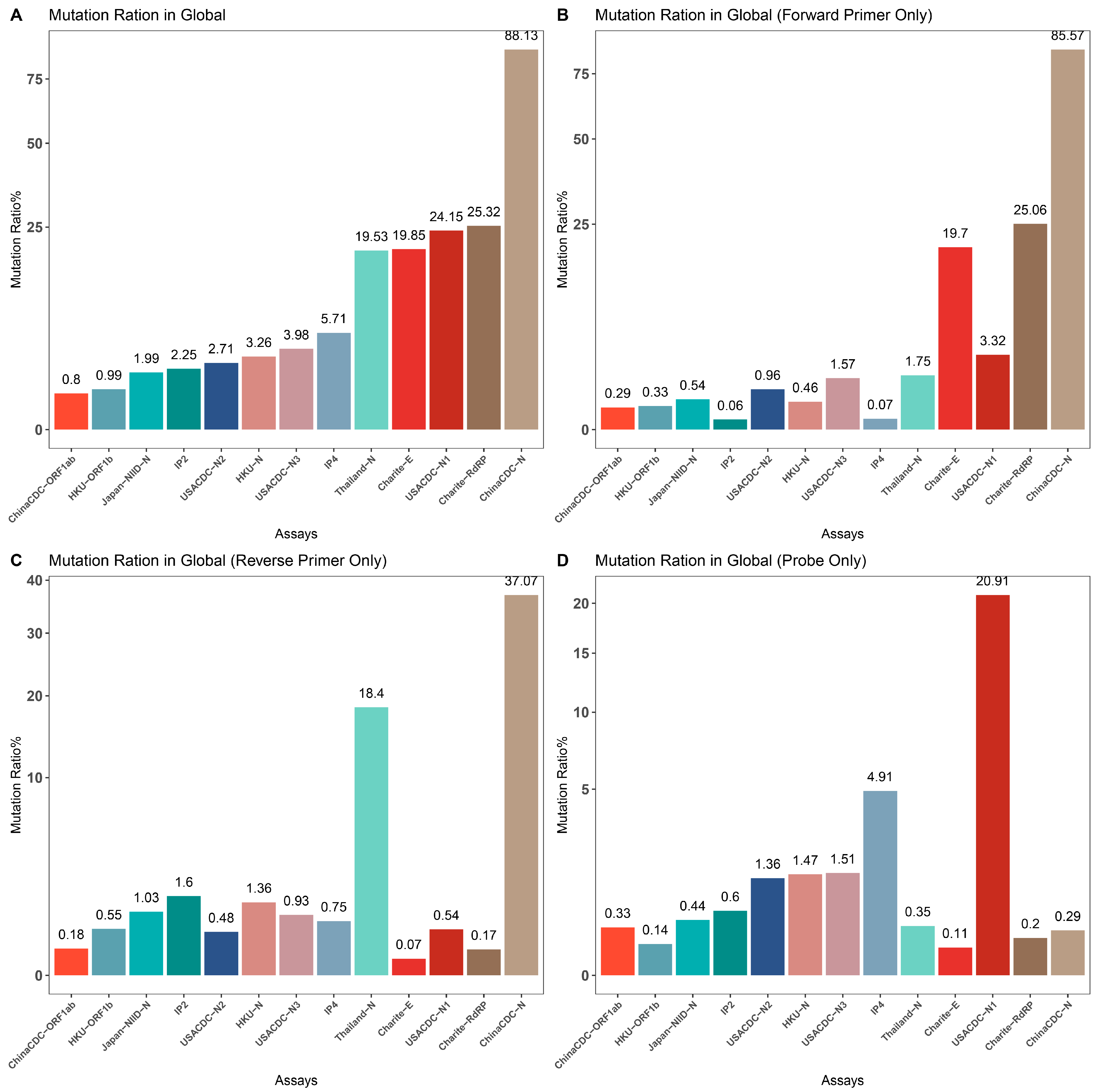
2.2.3. Assay Profile
2.2.4. Last Five Nucleotides of Primers
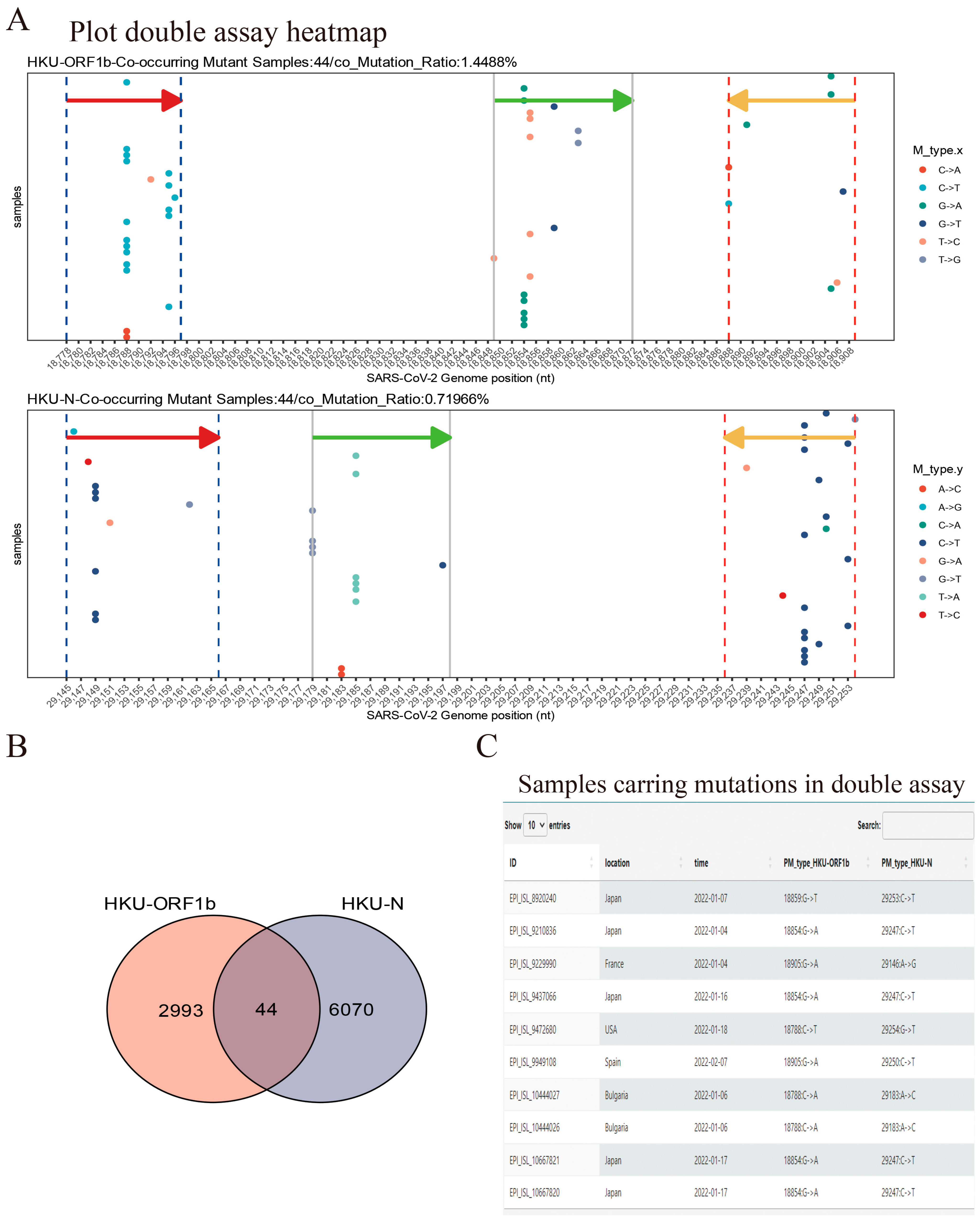
2.2.5. Double Assay Evaluation
2.2.6. RT-PCR Primer Design
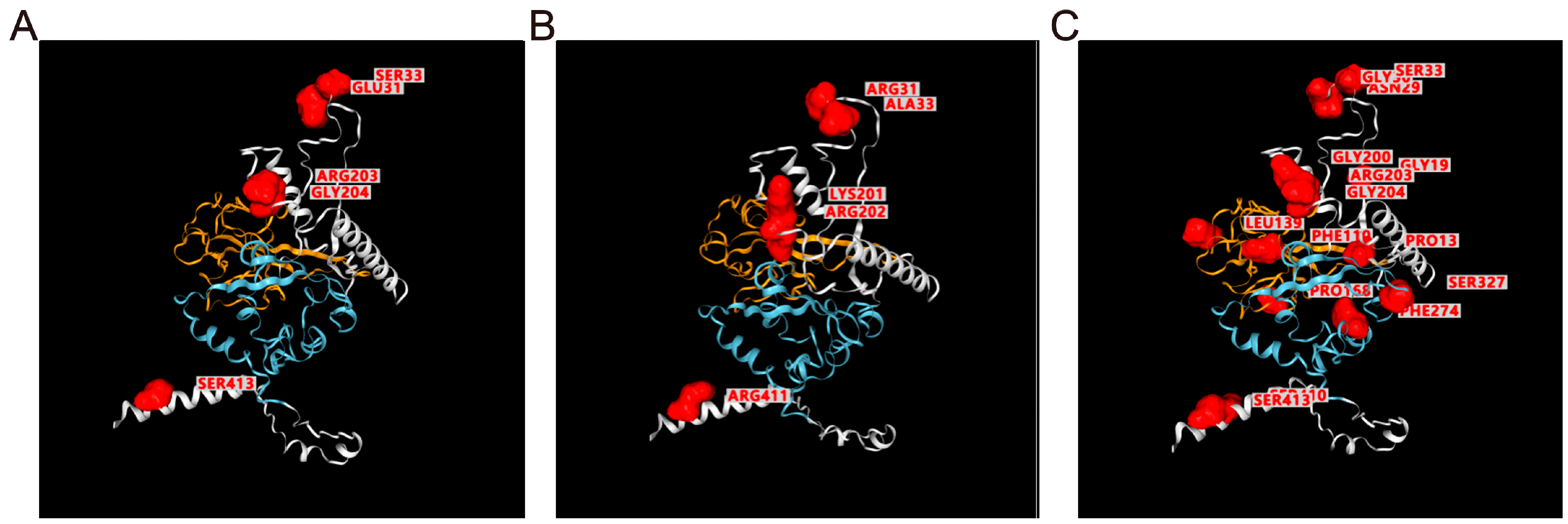
2.2.7. Structure Viewer for Mutated Proteins
3. Results
4. Discussions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO. Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 18 July 2023).
- Davis-Gardner, M.E.; Lai, L.; Wali, B.; Samaha, H.; Solis, D.; Lee, M.; Porter-Morrison, A.; Hentenaar, I.T.; Yamamoto, F.; Godbole, S.; et al. mRNA bivalent booster enhances neutralization against BA.2.75.2 and BQ.1.1. bioRxiv 2022. [Google Scholar] [CrossRef]
- Kurhade, C.; Zou, J.; Xia, H.; Liu, M.; Chang, H.C.; Ren, P.; Xie, X.; Shi, P.Y. Low neutralization of SARS-CoV-2 Omicron BA.2.75.2, BQ.1.1 and XBB.1 by parental mRNA vaccine or a BA.5 bivalent booster. Nat. Med. 2023, 29, 344–347. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.; Hachmann, N.P.; Collier, A.-r.Y.; Lasrado, N.; Mazurek, C.R.; Patio, R.C.; Powers, O.; Surve, N.; Theiler, J.; Korber, B.; et al. Substantial Neutralization Escape by SARS-CoV-2 Omicron Variants BQ.1.1 and XBB.1. N. Engl. J. Med. 2023, 388, 662–664. [Google Scholar] [CrossRef] [PubMed]
- Planas, D.; Bruel, T.; Staropoli, I.; Guivel-Benhassine, F.; Porrot, F.; Maes, P.; Grzelak, L.; Prot, M.; Mougari, S.; Planchais, C.; et al. Resistance of Omicron subvariants BA.2.75.2, BA.4.6 and BQ.1.1 to neutralizing antibodies. bioRxiv 2022. [Google Scholar] [CrossRef]
- Zou, J.; Kurhade, C.; Patel, S.; Kitchin, N.; Tompkins, K.; Cutler, M.; Cooper, D.; Yang, Q.; Cai, H.; Muik, A.; et al. Neutralization of BA.4–BA.5, BA.4.6, BA.2.75.2, BQ.1.1, and XBB.1 with Bivalent Vaccine. N. Engl. J. Med. 2023, 388, 854–857. [Google Scholar] [CrossRef]
- Jiang, X.L.; Zhu, K.L.; Wang, X.J.; Wang, G.L.; Li, Y.K.; He, X.J.; Sun, W.K.; Huang, P.X.; Zhang, J.Z.; Gao, H.X.; et al. Omicron BQ.1 and BQ.1.1 escape neutralisation by omicron subvariant breakthrough infection. Lancet Infect. Dis. 2023, 23, 28–30. [Google Scholar] [CrossRef]
- Li, D.; Duan, M.; Wang, X.; Gao, P.; Zhao, X.; Xu, K.; Gao, G.F. Neutralization of BQ.1, BQ.1.1, and XBB with RBD-Dimer Vaccines. N. Engl. J. Med. 2023, 388, 1142–1145. [Google Scholar] [CrossRef]
- Qu, P.; Evans, J.P.; Faraone, J.N.; Zheng, Y.M.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Lozanski, G.; et al. Enhanced neutralization resistance of SARS-CoV-2 Omicron subvariants BQ.1, BQ.1.1, BA.4.6, BF.7, and BA.2.75.2. Cell Host Microbe 2023, 31, 9–17.e13. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J.; et al. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 2023, 186, 279–286.e278. [Google Scholar] [CrossRef]
- Cao, Y.; Song, W.; Wang, L.; Liu, P.; Yue, C.; Jian, F.; Yu, Y.; Yisimayi, A.; Wang, P.; Wang, Y.; et al. Characterization of the enhanced infectivity and antibody evasion of Omicron BA.2.75. Cell Host Microbe 2022, 30, 1527–1539.e1525. [Google Scholar] [CrossRef]
- Yue, C.; Song, W.; Wang, L.; Jian, F.; Chen, X.; Gao, F.; Shen, Z.; Wang, Y.; Wang, X.; Cao, Y. ACE2 binding and antibody evasion in enhanced transmissibility of XBB.1.5. Lancet Infect. Dis. 2023, 23, 278–280. [Google Scholar] [CrossRef]
- World Health Organization. EG.5 Initial Risk Evaluation. Available online: https://www.who.int/docs/default-source/coronaviruse/09082023eg.5_ire_final.pdf (accessed on 21 September 2023).
- Liu, Y.; Liu, J.; Johnson, B.A.; Xia, H.; Ku, Z.; Schindewolf, C.; Widen, S.G.; An, Z.; Weaver, S.C.; Menachery, V.D.; et al. Delta spike P681R mutation enhances SARS-CoV-2 fitness over Alpha variant. Cell Rep. 2022, 39, 110829. [Google Scholar] [CrossRef] [PubMed]
- Parums, D.V. Editorial: A Rapid Global Increase in COVID-19 is Due to the Emergence of the EG.5 (Eris) Subvariant of Omicron SARS-CoV-2. Med. Sci. Monit. 2023, 29, e942244. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, F.; Ciccozzi, M. On the SARS-CoV-2 BA.2.86 lineage: A mutation point of view. J. Med. Virol. 2023, 95, e29079. [Google Scholar] [CrossRef] [PubMed]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Wu, T.; Hu, E.; Ge, X.; Yu, G. nCov2019: An R package for studying the COVID-19 coronavirus pandemic. PeerJ 2021, 9, e11421. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef]
- Portelli, S.; Olshansky, M.; Rodrigues, C.H.M.; D’Souza, E.N.; Myung, Y.; Silk, M.; Alavi, A.; Pires, D.E.V.; Ascher, D.B. Exploring the structural distribution of genetic variation in SARS-CoV-2 with the COVID-3D online resource. Nat. Genet. 2020, 52, 999–1001. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic. Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Gumna, J.; Antczak, M.; Adamiak, R.W.; Bujnicki, J.M.; Chen, S.-J.; Ding, F.; Ghosh, P.; Li, J.; Mukherjee, S.; Nithin, C.; et al. Computational Pipeline for Reference-Free Comparative Analysis of RNA 3D Structures Applied to SARS-CoV-2 UTR Models. Int. J. Mol. Sci. 2022, 23, 9630. [Google Scholar] [CrossRef] [PubMed]
- Rangan, R.; Watkins, A.M.; Chacon, J.; Kretsch, R.; Kladwang, W.; Zheludev, I.N.; Townley, J.; Rynge, M.; Thain, G.; Das, R. De novo 3D models of SARS-CoV-2 RNA elements from consensus experimental secondary structures. Nucleic. Acids Res. 2021, 49, 3092–3108. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Xu, Y.; Zheng, R.; Zeng, X.; Chen, Z.; Lin, S.; Xia, Z.; Liao, Y.; Zhang, Y.; Li, Q. Accessible and adaptable multiplexed real-time PCR approaches to identify SARS-CoV-2 variants of concern. Microbiol. Spectr. 2022, 10, e03222. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brunink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro. Surveill. 2020, 25, 2000045. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhang, J.; Li, J. Primer design for quantitative real-time PCR for the emerging Coronavirus SARS-CoV-2. Theranostics 2020, 10, 7150–7162. [Google Scholar] [CrossRef]
- Taiaroa, G.; Rawlinson, D.; Featherstone, L.; Pitt, M.; Caly, L.; Druce, J.; Purcell, D.; Harty, L.; Tran, T.; Roberts, J.; et al. Direct RNA sequencing and early evolution of SARS-CoV-2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- In Vitro Diagnostics EUAs. Available online: https://www.fda.gov/medical-devices/coronavirus-disease-2019-covid-19-emergency-use-authorizations-medical-devices/in-vitro-diagnostics-euas#individual-molecular (accessed on 6 March 2023).
- SARS-CoV-2 Reference Panel Comparative Data. Available online: https://www.fda.gov/medical-devices/coronavirus-covid-19-and-medical-devices/sars-cov-2-reference-panel-comparative-data (accessed on 6 March 2023).
- Rakotosamimanana, N.; Randrianirina, F.; Randremanana, R.; Raherison, M.S.; Rasolofo, V.; Solofomalala, G.D.; Spiegel, A.; Heraud, J.M. GeneXpert for the diagnosis of COVID-19 in LMICs. Lancet Glob. Health 2020, 8, e1457–e1458. [Google Scholar] [CrossRef]
- Woloshin, S.; Patel, N.; Kesselheim, A.S. False Negative Tests for SARS-CoV-2 Infection—Challenges and Implications. N. Engl. J. Med. 2020, 383, e38. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.C.; Bains, A.; Holland, L.A.; Smith, M.F.; Sullins, R.A.; Mellor, N.J.; Thomas, A.W.; Johnson, N.; Murugan, V.; Lim, E.S. SARS-CoV-2 Delta Variant N Gene Mutations Reduce Sensitivity to the TaqPath COVID-19 Multiplex Molecular Diagnostic Assay. Viruses 2022, 14, 1316. [Google Scholar] [CrossRef]
- Investigation of Novel SARS-CoV-2 Variant: Variant of Concern 202012/01: Technical Briefing Document on Novel SARS-CoV-2 Variant. Available online: https://www.gov.uk/government/publications/investigation-of-novel-sars-cov-2-variant-variant-of-concern-20201201 (accessed on 11 September 2023).
- Meng, B.; Kemp, S.A.; Papa, G.; Datir, R.; Ferreira, I.; Marelli, S.; Harvey, W.T.; Lytras, S.; Mohamed, A.; Gallo, G.; et al. Recurrent emergence of SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.1.7. Cell Rep. 2021, 35, 109292. [Google Scholar] [CrossRef] [PubMed]
- Kilic, T.; Weissleder, R.; Lee, H. Molecular and Immunological Diagnostic Tests of COVID-19: Current Status and Challenges. iScience 2020, 23, 101406. [Google Scholar] [CrossRef] [PubMed]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Bodenhofer, U.; Bonatesta, E.; Horejš-Kainrath, C.; Hochreiter, S. msa: An R package for multiple sequence alignment. Bioinformatics 2015, 31, 3997–3999. [Google Scholar] [CrossRef]
- Persson, S.; Larsson, C.; Simonsson, M.; Ellström, P. rprimer: An R/bioconductor package for design of degenerate oligos for sequence variable viruses. BMC Bioinform. 2022, 23, 239. [Google Scholar] [CrossRef]
- Guex, N.; Peitsch, M.C.; Schwede, T. Automated comparative protein structure modeling with SWISS-MODEL and Swiss-PdbViewer: A historical perspective. Electrophoresis 2009, 30 (Suppl. S1), S162–S173. [Google Scholar] [CrossRef]
- Waterhouse, A.; Bertoni, M.; Bienert, S.; Studer, G.; Tauriello, G.; Gumienny, R.; Heer, F.T.; de Beer, T.A.P.; Rempfer, C.; Bordoli, L.; et al. SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic. Acids Res. 2018, 46, W296–W303. [Google Scholar] [CrossRef]
- Zhou, X.; Zheng, W.; Li, Y.; Pearce, R.; Zhang, C.; Bell, E.W.; Zhang, G.; Zhang, Y. I-TASSER-MTD: A deep-learning-based platform for multi-domain protein structure and function prediction. Nat. Protoc. 2022, 17, 2326–2353. [Google Scholar] [CrossRef]
- Wilhelm, A.; Toptan, T.; Pallas, C.; Wolf, T.; Goetsch, U.; Gottschalk, R.; Vehreschild, M.J.G.T.; Ciesek, S.; Widera, M. Antibody-Mediated Neutralization of Authentic SARS-CoV-2 B.1.617 Variants Harboring L452R and T478K/E484Q. Viruses 2021, 13, 1693. [Google Scholar] [CrossRef] [PubMed]
- Zou, J.; Xie, X.; Fontes-Garfias, C.R.; Swanson, K.A.; Kanevsky, I.; Tompkins, K.; Cutler, M.; Cooper, D.; Dormitzer, P.R.; Shi, P.-Y. The effect of SARS-CoV-2 D614G mutation on BNT162b2 vaccine-elicited neutralization. Npj. Vaccines. 2021, 6, 44. [Google Scholar] [CrossRef] [PubMed]
- Callaway, E. The coronavirus is mutating—Does it matter? Nature 2020, 585, 174–177. [Google Scholar] [CrossRef] [PubMed]
- Diagnostic Detection of Novel Coronavirus 2019 by Real Time RT-PCR. Available online: https://www.who.int/docs/default-source/coronaviruse/conventional-rt-pcr-followed-by-sequencing-for-detection-of-ncov-rirl-nat-inst-health-t.pdf?sfvrsn=42271c6d_4 (accessed on 21 September 2023).
- Real-Time RT-PCR Assays for the Detection of SARS-CoV-2. Available online: https://www.who.int/docs/default-source/coronaviruse/real-time-rt-pcr-assays-for-the-detection-of-sars-cov-2-institut-pasteur-paris.pdf?sfvrsn=3662fcb6_2 (accessed on 21 September 2023).
- Shirato, K.; Nao, N.; Katano, H.; Takayama, I.; Saito, S.; Kato, F.; Katoh, H.; Sakata, M.; Nakatsu, Y.; Mori, Y.; et al. Development of Genetic Diagnostic Methods for Detection for Novel Coronavirus 2019 (nCoV-2019) in Japan. Jpn J. Infect. Dis. 2020, 73, 304–307. [Google Scholar] [CrossRef]
- 2019-Novel Coronavirus (2019-nCoV) Real-Time rRT-PCR Panel Primers and Probes. Available online: https://stacks.cdc.gov/view/cdc/84525 (accessed on 21 September 2023).
- Chu, D.K.W.; Pan, Y.; Cheng, S.M.S.; Hui, K.P.Y.; Krishnan, P.; Liu, Y.; Ng, D.Y.M.; Wan, C.K.C.; Yang, P.; Wang, Q.; et al. Molecular Diagnosis of a Novel Coronavirus (2019-nCoV) Causing an Outbreak of Pneumonia. Clin. Chem. 2020, 66, 549–555. [Google Scholar] [CrossRef]
- Specific Primers and Probes for Detection 2019 Novel Coronavirus. Available online: https://ivdc.chinacdc.cn/kyjz/202001/t20200121_211337.html (accessed on 21 September 2023).
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Gu, Z.; Gu, L.; Eils, R.; Schlesner, M.; Brors, B. circlize Implements and enhances circular visualization in R. Bioinformatics 2014, 30, 2811–2812. [Google Scholar] [CrossRef] [PubMed]
- Stadhouders, R.; Pas, S.D.; Anber, J.; Voermans, J.; Mes, T.H.; Schutten, M. The effect of primer-template mismatches on the detection and quantification of nucleic acids using the 5' nuclease assay. J. Mol. Diagn. 2010, 12, 109–117. [Google Scholar] [CrossRef]
- Vanaerschot, M.; Mann, S.A.; Webber, J.T.; Kamm, J.; Bell, S.M.; Bell, J.; Hong, S.N.; Nguyen, M.P.; Chan, L.Y.; Bhatt, K.D.; et al. Identification of a Polymorphism in the N Gene of SARS-CoV-2 That Adversely Impacts Detection by Reverse Transcription-PCR. J. Clin. Microbiol. 2020, 59. [Google Scholar] [CrossRef]
- He, Y.; Zhou, Y.; Liu, S.; Kou, Z.; Li, W.; Farzan, M.; Jiang, S. Receptor-binding domain of SARS-CoV spike protein induces highly potent neutralizing antibodies: Implication for developing subunit vaccine. Biochem. Biophys. Res. Commun. 2004, 324, 773–781. [Google Scholar] [CrossRef] [PubMed]
- CDC. Coronavirus Disease 2019 (COVID-19). Centers for Disease Control and Prevention. Available online: https://www.cdc.gov/coronavirus/2019-ncov/index.html (accessed on 21 September 2023).



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ma, S.; Xiao, G.; Deng, X.; Tong, M.; Huang, J.; Li, Q.; Zhang, Y. CovidShiny: An Integrated Web Tool for SARS-CoV-2 Mutation Profiling and Molecular Diagnosis Assay Evaluation In Silico. Viruses 2023, 15, 2017. https://doi.org/10.3390/v15102017
Ma S, Xiao G, Deng X, Tong M, Huang J, Li Q, Zhang Y. CovidShiny: An Integrated Web Tool for SARS-CoV-2 Mutation Profiling and Molecular Diagnosis Assay Evaluation In Silico. Viruses. 2023; 15(10):2017. https://doi.org/10.3390/v15102017
Chicago/Turabian StyleMa, Shaoqian, Gezhi Xiao, Xusheng Deng, Mengsha Tong, Jialiang Huang, Qingge Li, and Yongyou Zhang. 2023. "CovidShiny: An Integrated Web Tool for SARS-CoV-2 Mutation Profiling and Molecular Diagnosis Assay Evaluation In Silico" Viruses 15, no. 10: 2017. https://doi.org/10.3390/v15102017
APA StyleMa, S., Xiao, G., Deng, X., Tong, M., Huang, J., Li, Q., & Zhang, Y. (2023). CovidShiny: An Integrated Web Tool for SARS-CoV-2 Mutation Profiling and Molecular Diagnosis Assay Evaluation In Silico. Viruses, 15(10), 2017. https://doi.org/10.3390/v15102017