
Omicron Coronavirus: pH-Dependent Electrostatic Potential and Energy of Association of Spike Protein to ACE2 Receptor


Abstract
:1. Introduction
2. Methods
2.1. Protein Electrostatics
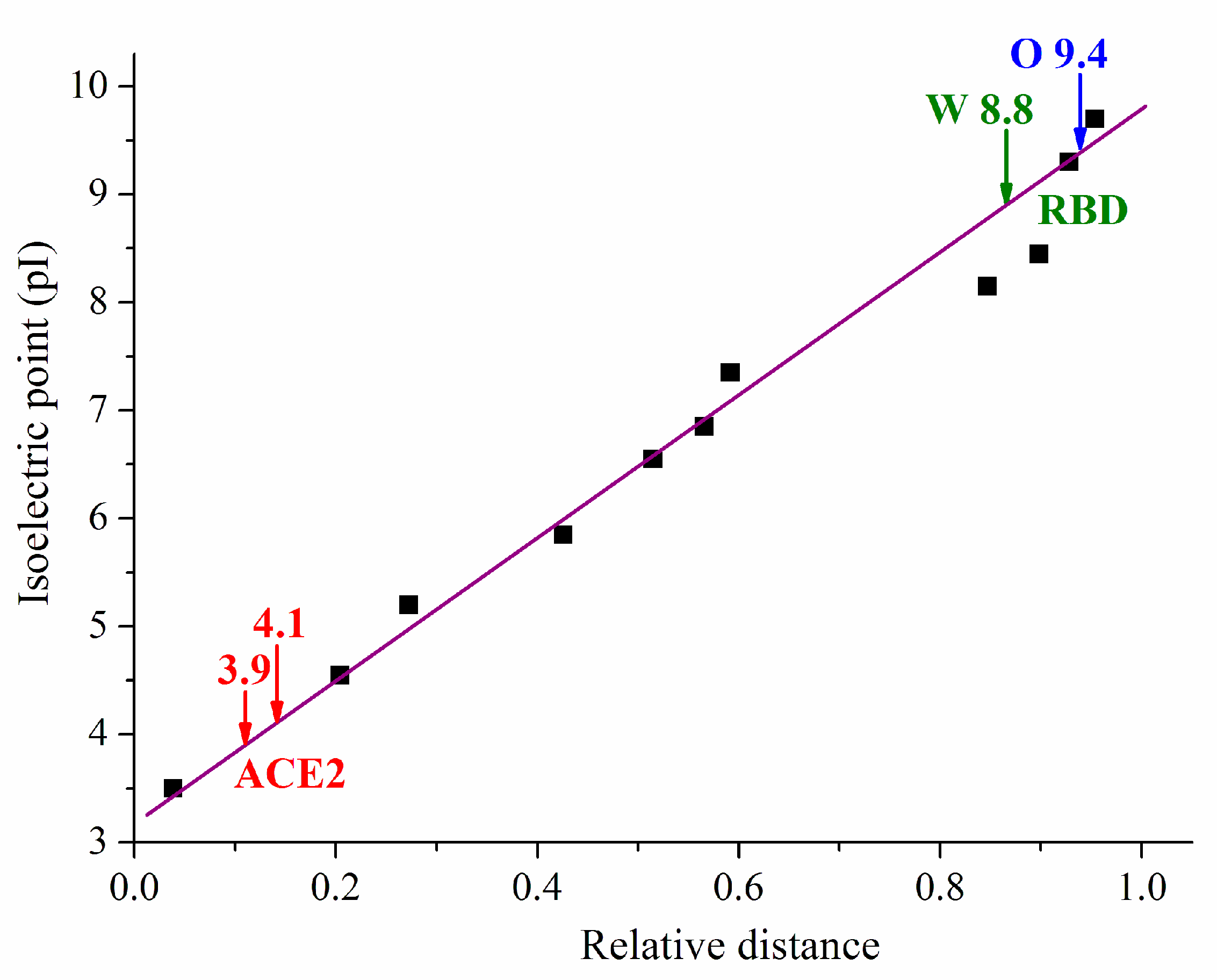
2.2. Isoelectric Focusing
3. Results
3.1. Molecular Models
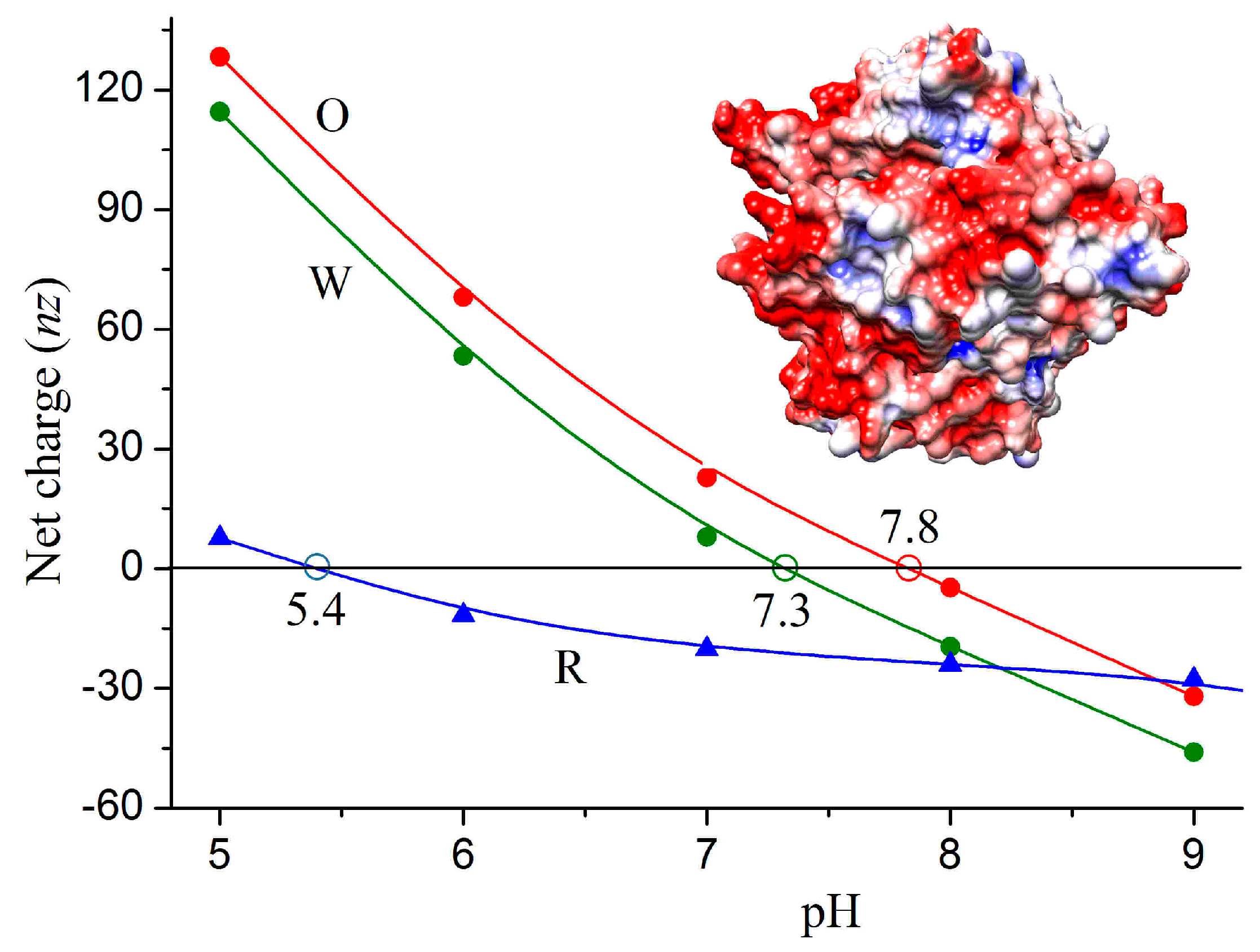
3.2. Association Energy
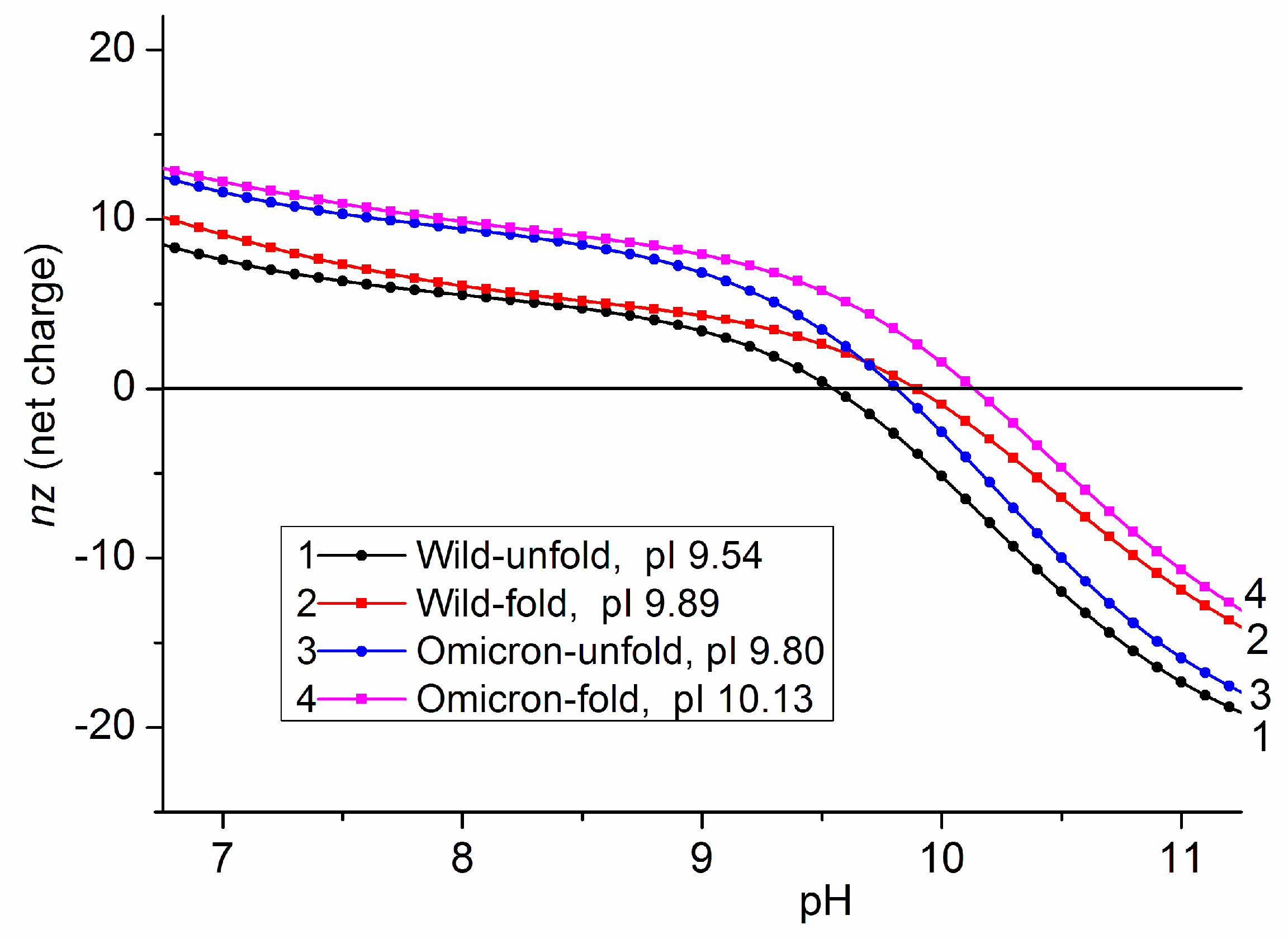
3.3. Isoelectric Points of 3D Protein Structures
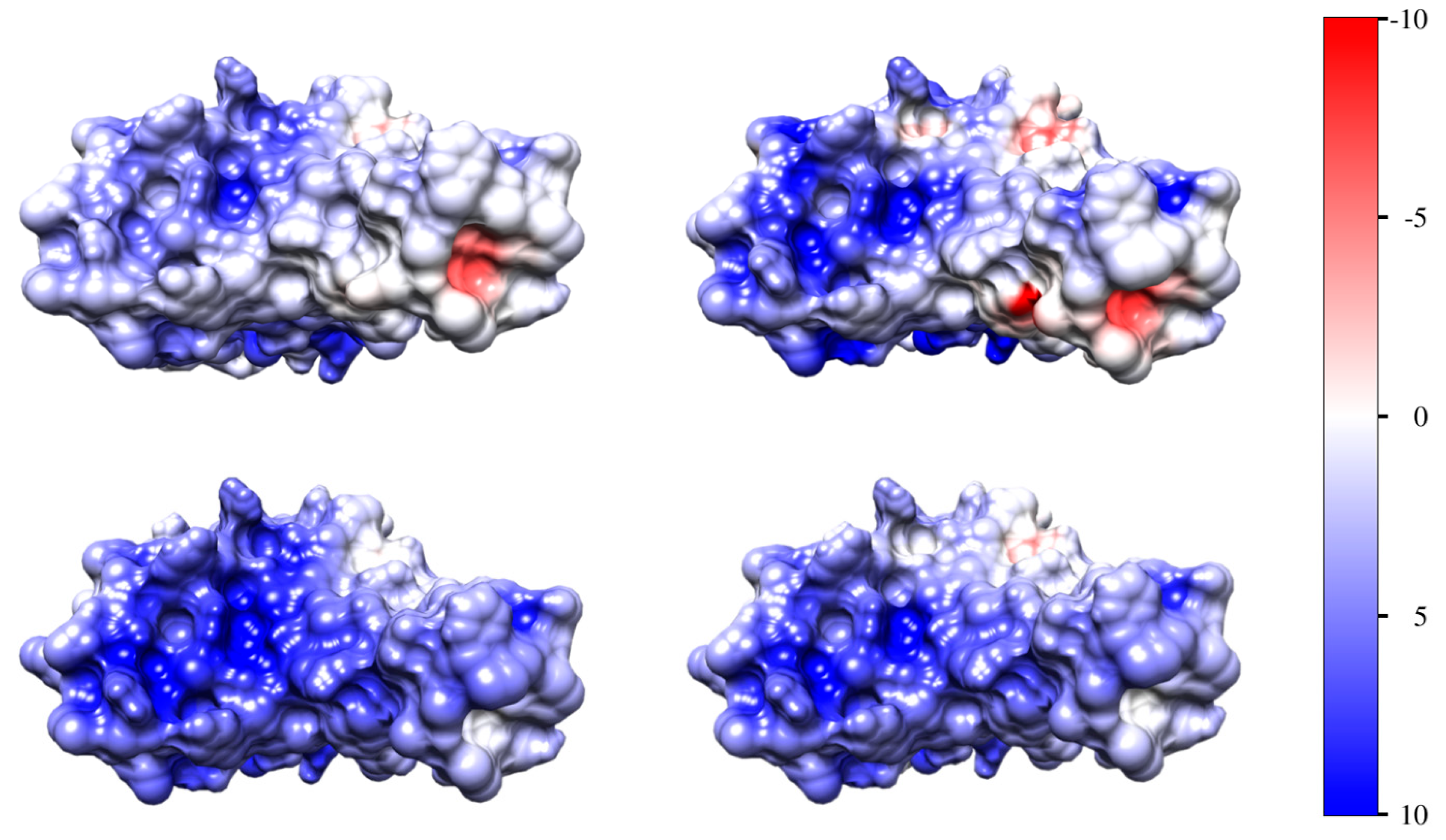
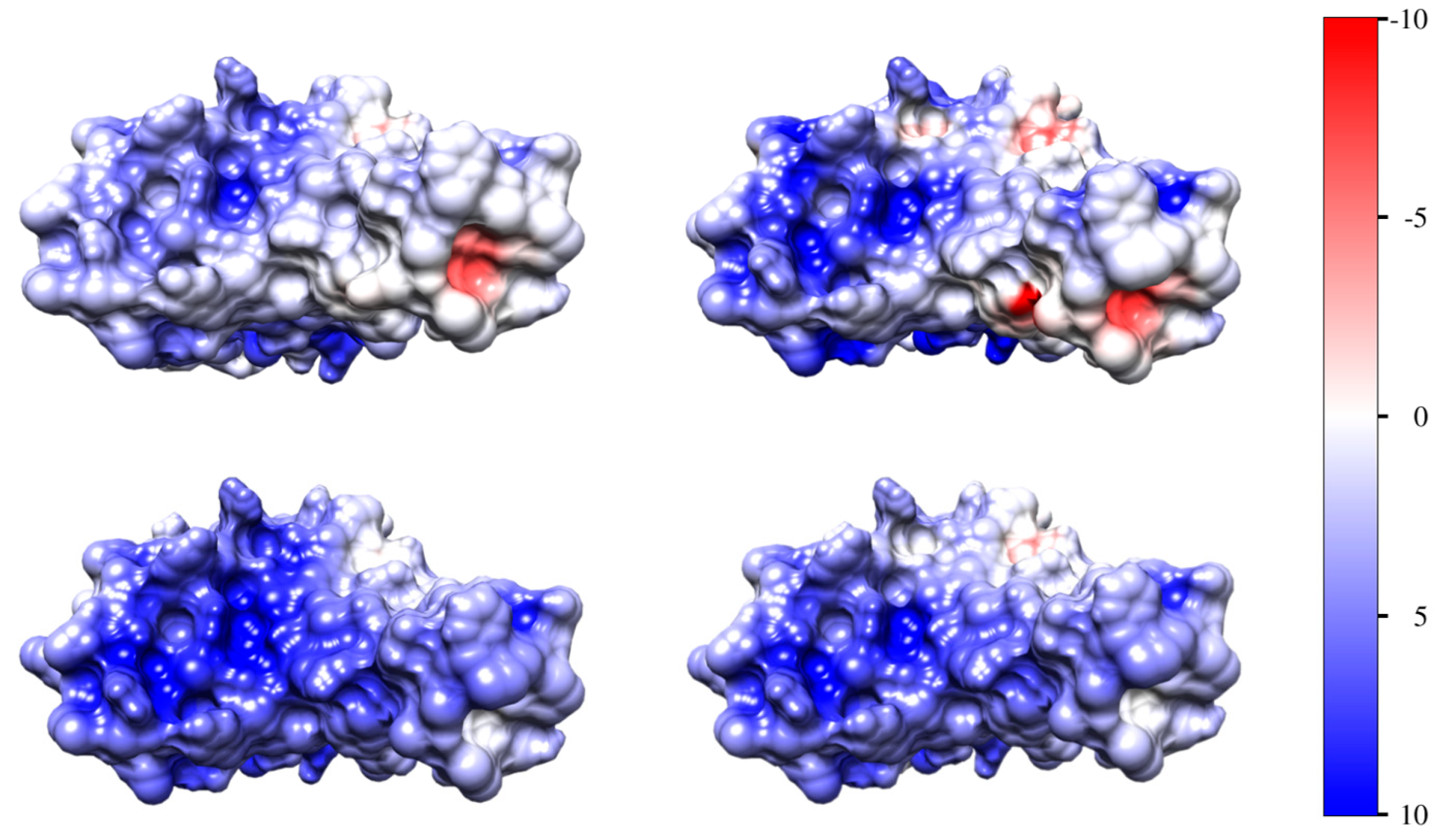
3.4. Surface Electrostatic Potential
3.5. Isoelectric Points of Unfolded Polypeptide Chains
4. Discussion
4.1. Energy of Association
4.2. Isoelectric Point and Surface Potential
4.3. Shift of the Isoelectric Point
4.4. Heterogeneity of the Electrostatic Potential
4.5. Recombinant Proteins and Posttranslational Modifications
4.6. Contagiousness and Pathogenicity
4.7. Comparison with Literature Data
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Classification of Omicron (B.1.1.529): SARS-CoV-2 Variant of Concern. Available online: https://www.who.int/news/item/26-11-2021-classification-of-omicron-(b.1.1.529)-sars-cov-2-variant-of-concern (accessed on 26 November 2021).
- Madhi, S.A.; Kwatra, G.; Myers, J.E.; Jassat, W.; Dhar, N.; Mukendi, C.K.; Nana, A.J.; Blumberg, L.; Welch, R.; Ngorima-Mabhena, N.; et al. Population Immunity and COVID-19 Severity with Omicron Variant in South Africa. N. Engl. J. Med. 2022, 386, 1314–1326. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Thambiraja, T.S.; Karuppanan, K.; Subramaniam, G. Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein. J. Med. Virol. 2021, 94, 1641–1649. [Google Scholar] [CrossRef] [PubMed]
- Bittmann, S. Role of Omicron variant of SARS-CoV-2 in children in Germany. World J. Pediatr. 2022, 18, 283–284. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Gilby, N.B.; Wei, G.W. Omicron variant (B.1.1.529): Infectivity, vaccine breakthrough, and antibody resistance. J. Chem. Inf. Model. 2022, 62, 412–422. [Google Scholar] [CrossRef]
- Tian, D.; Sun, Y.; Xu, H.; Ye, Q. The emergence and epidemic characteristics of the highly mutated SARS-CoV-2 Omicron variant. J. Med. Virol. 2022, 94, 2376–2383. [Google Scholar] [CrossRef]
- Maslo, C.; Friedland, R.; Toubkin, M.; Laubscher, A.; Akaloo, T.; Kama, B. Characteristics and outcomes of hospitalized patients in South Africa during the COVID-19 Omicron wave compared with previous waves. JAMA 2022, 327, 583–584. [Google Scholar] [CrossRef] [PubMed]
- Hardenbrook, N.J.; Zhang, P. A structural view of the SARS-CoV-2 virus and its assembly. Curr. Opin. Virol. 2022, 52, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yang, C.; Xu, X.F.; Xu, W.; Liu, S.W. Structural and functional properties of SARS-CoV-2 spike protein: Potential antivirus drug development for COVID-19. Acta Pharmacol. Sin. 2020, 41, 1141–1149. [Google Scholar] [CrossRef]
- Wang, Q.; Zhang, Y.; Wu, L.; Niu, S.; Song, C.; Zhang, Z.; Lu, G.; Qiao, C.; Hu, Y.; Yuen, K.Y.; et al. Structural and functional basis of SARS-CoV-2 entry by using human ACE2. Cell 2020, 181, 894–904. [Google Scholar] [CrossRef]
- Ortega, J.T.; Jastrzebska, B.; Rangel, H.R. Omicron SARS-CoV-2 variant spike protein shows an increased affinity to the human ACE2 receptor: An in silico analysis. Pathogens 2022, 11, 45. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Karki, C.B.; Du, D.; Li, H.; Wang, J.; Sobitan, A.; Teng, S.; Tang, Q.; Li, L. Spike proteins of SARS-CoV and SARS-CoV-2 utilize different mechanisms to bind with human ACE2. Front. Mol. Biosci. 2020, 7, 392. [Google Scholar] [CrossRef]
- Ali, F.; Elserafy, M.; Alkordi, M.H.; Amin, M. ACE2 coding variants in different populations and their potential impact on SARS-CoV-2 binding affinity. Biochem. Biophys. Rep. 2020, 24, 100798. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.Y. Key interacting residues between RBD of SARS-CoV-2 and ACE2 receptor: Combination of molecular dynamics simulation and density functional calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.Y. Binding interactions between receptor-binding domain of spike protein and human angiotensin converting enzyme-2 in omicron variant. J. Phys. Chem. Lett. 2022, 13, 3915–3921. [Google Scholar] [CrossRef]
- Xie, Y.; Guo, W.; Lopez-Hernadez, A.; Teng, S.; Li, L. The pH effects on SARS-CoV and SARS-CoV-2 spike proteins in the process of binding to hACE2. Pathogens 2022, 11, 238. [Google Scholar] [CrossRef]
- Javidpour, L.; Božič, A.; Naji, A.; Podgornik, R. Electrostatic interactions between the SARS-CoV-2 virus and a charged electret fibre. Soft Matter 2021, 17, 4296–4303. [Google Scholar] [CrossRef]
- Sang, P.; Chen, Y.Q.; Liu, M.T.; Wang, Y.T.; Yue, T.; Li, Y.; Yin, Y.R.; Yang, L.Q. Electrostatic interactions are the primary determinant of the binding affinity of SARS-CoV-2 spike RBD to ACE2: A computational case study of omicron variants. Int. J. Mol. Sci. 2022, 23, 14796. [Google Scholar]
- Pascarella, S.; Ciccozzi, M.; Zella, D.; Bianchi, M.; Benedetti, F.; Benvenuto, D.; Broccolo, F.; Cauda, R.; Caruso, A.; Angeletti, S.; et al. SARS-CoV-2 B.1.617 Indian variants: Are electrostatic potential changes responsible for a higher transmission rate? J. Med. Virol. 2021, 93, 6551–6556. [Google Scholar] [CrossRef]
- Pascarella, S.; Ciccozzi, M.; Bianchi, M.; Benvenuto, D.; Cauda, R.; Cassone, A. The value of electrostatic potentials of the spike receptor binding and N-terminal domains in addressing transmissibility and infectivity of SARS-CoV-2 variants of concern. J. Infect. 2022, 84, 62–63. [Google Scholar] [CrossRef] [PubMed]
- Nie, C.; Sahoo, A.K.; Netz, R.R.; Herrmann, A.; Ballauff, M.; Haag, R. Charge matters: Mutations in Omicron Variant Favor Binding to Cells. Chembiochem 2022, 23, e202100681. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, T.; Ozono, H.; Akisawa, K.; Hatada, R.; Okuwaki, K.; Mochizuki, Y. Interaction analysis on the SARS-CoV-2 Spike protein receptor binding domain using visualization of the interfacial electrostatic complementarity. J. Phys. Chem. Lett. 2021, 12, 11267–11272. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Xiong, D.; Luo, S.; Duan, L. Origin of the tight binding mode to ACE2 triggered by multi-point mutations in the omicron variant: A dynamic insight. Phys. Chem. Chem. Phys. 2022, 24, 8724–8737. [Google Scholar] [CrossRef] [PubMed]
- Goher, S.S.; Ali, F.; Amin, M. The Delta variant mutations in the receptor binding domain of SARS-CoV-2 show enhanced electrostatic interactions with the ACE2. Med. Drug Discov. 2022, 13, 100114. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Bhattacharyya, R.; Sengupta, J. Dynamics and electrostatics define an allosteric druggable site within the receptor-binding domain of SARS-CoV-2 spike protein. FEBS Lett. 2021, 595, 442–451. [Google Scholar] [CrossRef]
- Rath, S.L.; Padhi, A.K.; Mandal, N. Scanning the RBD-ACE2 molecular interactions in Omicron variant. Biochem. Biophys. Res. Commun. 2022, 592, 18–23. [Google Scholar] [CrossRef]
- Fazekas, Z.; Menyhárd, D.K.; Perczel, A. Omicron binding mode: Contact analysis and dynamics of the omicron receptor-binding domain in complex with ACE2. J. Chem. Inf. Model. 2022, 62, 3844–3853. [Google Scholar] [CrossRef]
- Lupala, C.S.; Ye, Y.; Chen, H.; Su, X.D.; Liu, H. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem. Biophys. Res. Commun. 2022, 590, 34–41. [Google Scholar] [CrossRef]
- Giron, C.C.; Laaksonen, A.; Barroso da Silva, F.L. Differences between Omicron SARS-CoV-2 RBD and other variants in their ability to interact with cell receptors and monoclonal antibodies. J. Biomol. Struct. Dyn. 2023, 41, 5707–5727. [Google Scholar] [CrossRef]
- Gan, H.H.; Zinno, J.; Piano, F.; Gunsalus, K.C. Omicron Spike protein has a positive electrostatic surface that promotes ACE2 recognition and antibody escape. Front. Virol. 2022, 23, 14796. [Google Scholar] [CrossRef]
- Shah, M.; Ahmad, B.; Choi, S.; Woo, H.G. Mutations in the SARS-CoV-2 spike RBD are responsible for stronger ACE2 binding and poor anti-SARS-CoV mAbs cross-neutralization. Comput. Struct. Biotechnol. J. 2022, 18, 3402–3414. [Google Scholar] [CrossRef] [PubMed]
- Dung, D.N.; Phan, A.D.; Nguyen, T.T.; Lam, V.D. Effects of surface charge and environmental factors on the electrostatic interaction of fiber with virus-like particle: A case of coronavirus. AIP Adv. 2021, 11, 10508. [Google Scholar]
- Barroso da Silva, F.L.; Giron, C.C.; Laaksonen, A. Electrostatic features for the receptor binding domain of SARS-COV-2 wildtype and its variants. Compass to the severity of the future variants with the charge-rule. J. Phys. Chem. B 2022, 126, 6835–6852. [Google Scholar] [CrossRef] [PubMed]
- Aksenova, A.Y.; Likhachev, I.V.; Grishin, S.Y.; Galzitskaya, O.V. The Increased Amyloidogenicity of Spike RBD and pH-Dependent Binding to ACE2 May Contribute to the Transmissibility and Pathogenic Properties of SARS-CoV-2 Omicron as Suggested by In Silico Study. Int. J. Mol. Sci. 2022, 23, 13502. [Google Scholar] [CrossRef]
- Sevenich, M.; van den Heuvel, J.; Gering, I.; Mohrlüder, J.; Willbold, D. A so-far overlooked secondary conformation state in the binding mode of SARS-CoV-2 spike protein to human ACE2 and its conversion rate are crucial for estimating infectivity efficacy of the underlying virus variant. J. Virol. 2022, 96, e00685-22. [Google Scholar] [CrossRef]
- Basavarajappa, S.C.; Liu, A.R.; Bruchez, A.; Li, Z.; Suzart, V.G.; Liu, Z.; Chen, Y.; Xiao, T.S.; Buck, M.; Ramakrishnan, P. Trimeric receptor-binding domain of SARS-CoV-2 acts as a potent inhibitor of ACE2 receptor-mediated viral entry. iScience 2022, 25, 104716. [Google Scholar] [CrossRef]
- Pawłowski, P. SARS-CoV-2 variant Omicron (B.1.1.529) is in a rising trend of mutations increasing the positive electric charge in crucial regions of the spike protein S. Acta Biochim. Pol. 2022, 69, 263–264. [Google Scholar] [CrossRef]
- Hanai, T. Quantitative in silico analysis of SARS-CoV-2 S-RBD omicron mutant transmissibility. Talanta 2022, 240, 123206. [Google Scholar] [CrossRef]
- Kumar, R.; Murugan, N.A.; Srivastava, V. Improved binding affinity of omicron’s spike protein for the human angiotensin-converting enzyme 2 receptor is the key behind its increased virulence. Int. J. Mol. Sci. 2022, 23, 3409. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C.; et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Liu, P.; Wang, N.; Wang, L.; Fan, K.; Zhu, Q.; Wang, K.; Chen, R.; Feng, R.; Jia, Z.; et al. Structural and functional characterizations of infectivity and immune evasion of SARS-CoV-2 Omicron. Cell 2022, 185, 860–871. [Google Scholar] [CrossRef]
- Shishir, T.A.; Jannat, T.; Naser, I.B. An in-silico study of the mutation-associated effects on the spike protein of SARS-CoV-2, Omicron variant. PLoS ONE 2022, 17, e0266844. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Wang, Y.; Liu, C.; Zhang, C.; Han, W.; Hong, X.; Wang, Y.; Hong, Q.; Wang, S.; Zhao, Q.; et al. Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM. Sci. Adv. 2021, 7, eabe5575. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Im, W.; Beglov, D.; Roux, B. Continuum solvation model: Computation of electrostatic forces from numerical solutions to the Poisson-Boltzmann equation. Comput. Phys. Commun. 1998, 111, 59–75. [Google Scholar] [CrossRef]
- Jo, S.; Vargyas, M.; Vasko-Szedlar, J.; Roux, B.; Im, W. PBEQ-Solver for online visualization of electrostatic potential of biomolecules. Nucleic Acids Res. 2008, 36, W270–W275. [Google Scholar] [CrossRef]
- Pandurangan, A.P.; Ochoa-Montano, B.; Ascher, D.B.; Blundell, T.L. SDM: A server for predicting effects of mutations on protein stability. Nucleic Acids Res. 2017, 45, W229–W235. [Google Scholar] [CrossRef]
- Worth, C.L.; Preissner, R.; Blundell, T.L. SDM—A server for predicting effects of mutations on protein stability and malfunction. Nucleic Acids Res. 2011, 39 (Suppl. S2), W215–W222. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Li, H.; Robertson, A.D.; Jensen, J.H. Very fast empirical prediction and rationalization of protein pKa values. Proteins Struct. Funct. Bioinform. 2005, 61, 704–721. [Google Scholar] [CrossRef] [PubMed]
- Jurrus, E.; Engel, D.; Star, K.; Monson, K.; Brandi, J.; Felberg, L.E.; Brookes, D.H.; Wilson, L.; Chen, J.; Liles, K.; et al. Improvements to the APBS biomolecular solvation software suite. Protein Sci. 2018, 27, 112–128. [Google Scholar] [CrossRef] [PubMed]
- Walsh, I.; Minervini, G.; Corazza, A.; Esposito, G.; Tosatto, S.C.; Fogolari, F. Bluues server: Electrostatic properties of wild-type and mutated protein structures. Bioinformatics 2012, 28, 2189–2190. [Google Scholar] [CrossRef] [PubMed]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Wu, G.; Chen, Q.; Yu, C.; Xu, G.; Liu, A.; Wang, L. Fingerprinting trimeric SARS-CoV-2 RBD by capillary isoelectric focusing with whole-column imaging detection. Anal. Biochem. 2023, 663, 115034. [Google Scholar] [CrossRef]
- Calibration Kits for pI Deteminations Using Isoelectric Focusing; GE Healthcare Life Sciences: Amersham, UK, 2013.
- Hristova, S.H.; Zhivkov, A.M. Isoelectric point of free and adsorbed cytochrome c determined by various methods. Colloids Surf. B Biointerfaces 2019, 174, 87–94. [Google Scholar] [CrossRef]
- Hristova, S.; Zhivkov, A. Electrooptical determination of the isoelectric point of globular proteins: Cytochrome c adsorbed on montmorillonite nanoplates. Colloids Surf. B Biointerfaces 2019, 176, 480–487. [Google Scholar] [CrossRef]
- Benton, D.J.; Wrobel, A.G.; Xu, P.; Roustan, C.; Martin, S.R.; Rosenthal, P.B.; Skehel, J.J.; Gamblin, S.J. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature 2020, 588, 327–330. [Google Scholar] [CrossRef]
- Xiong, X.; Qu, K.; Ciazynska, K.A.; Hosmillo, M.; Carter, A.P.; Ebrahimi, S.; Ke, Z.; Scheres, S.H.; Bergamaschi, L.; Grice, G.L.; et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat. Struct. Mol. Biol. 2020, 27, 934–941. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Ye, F.; Guo, Y.; Xia, L.; Zhong, X.; Chi, X.; Zhou, Q. Structural basis for the different states of the spike protein of SARS-CoV-2 in complex with ACE2. Cell Res. 2021, 31, 717–719. [Google Scholar] [CrossRef]
- Pramanick, I.; Sengupta, N.; Mishra, S.; Pandey, S.; Girish, N.; Das, A.; Dutta, S. Conformational flexibility and structural variability of SARS-CoV2 S-protein. Structure 2021, 29, 834–845. [Google Scholar] [CrossRef]
- Panthi, B.; Dutta, S.; Chandra, A. All-atom simulations of the trimeric spike protein of SARS-CoV-2 in aqueous medium: Nature of interactions, conformational stability and free energy diagrams for conformational transition of the protein. J. Comput. Chem. 2023, 44, 1560–1577. [Google Scholar] [CrossRef]
- Ke, Z.; Oton, J.; Qu, K.; Cortese, M.; Zila, V.; McKeane, L.; Nakane, T.; Zivanov, J.; Neufeldt, C.J.; Cerikan, B.; et al. Structures and distributions of SARS-CoV-2 spike proteins on intact virions. Nature 2020, 588, 498–502. [Google Scholar] [CrossRef] [PubMed]
- Lobo, V.R.; Warwicker, J. Predicted pH-dependent stability of SARS-CoV-2 spike protein trimer from interfacial acidic groups. Comput. Struct. Biotechnol. J. 2021, 19, 5140–5148. [Google Scholar] [CrossRef]
- Conroy, J.M.; Hartley, J.L.; Soffer, R.L. Canine pulmonary angiotensin-converting enzyme. Physicochemical, catalytic and immunological properties. Biochim. Biophys. Acta (BBA)-Enzymol. 1978, 524, 403–412. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A human homolog of angiotensin-converting enzyme: Cloning and functional expression as a captopril-insensitive carboxypeptidase. J. Biol. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef]
- Bünning, P.; Budek, W.; Escher, R.; Schönherr, E. Characteristics of angiotensin converting enzyme and its role in the metabolism of angiotensin I by endothelium. J. Cardiovasc. Pharmacol. 1986, 8, S52–S57. [Google Scholar] [CrossRef] [PubMed]
- Lanzillo, J.J.; Stevens, J.; Dasarathy, Y.; Yotsumoto, H.; Fanburg, B.L. Angiotensin-converting enzyme from human tissues. Physicochemical, catalytic, and immunological properties. J. Biol. Chem. 1985, 260, 14938–14944. [Google Scholar] [CrossRef] [PubMed]
- Lanzillo, J.J.; Joanne, S.; Tumas, J.; Fanburg, B.L. Spontaneous change of human plasma angiotensin I converting enzyme isoelectric point. Arch. Biochem. Biophys. 1983, 227, 434–439. [Google Scholar] [CrossRef]
- Houston, M.C. Pleural Fluid PH: Diagnostic, therapeutic, and prognostic value. Am. J. Surg. 1987, 154, 333–337. [Google Scholar] [CrossRef] [PubMed]
- Fischer, H.; Widdicombe, J.H. Mechanisms of acid and base secretion by the airway epithelium. J. Membr. Biol. 2006, 211, 139. [Google Scholar] [CrossRef] [PubMed]
- Tosta, S.; Moreno, K.; Schuab, G.; Fonseca, V.; Segovia, F.M.C.; Kashima, S.; Elias, M.C.; Sampaio, S.C.; Ciccozzi, M.; Alcantara, L.C.J.; et al. Global SARS-CoV-2 genomic surveillance: What we have learned (so far). Infect. Genet. Evol. 2023, 108, 105405. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Jian, F.; Wang, J.; Yu, Y.; Song, W.; Yisimayi, A.; Wang, J.; An, R.; Chen, X.; Zhang, N.; et al. Imprinted SARS-CoV-2 humoral immunity induces convergent Omicron RBD evolution. Nature 2023, 614, 521–529. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, F.; Sanna, D.; Azzena, I.; Casu, M.; Cossu, P.; Fiori, P.L.; Benvenuto, D.; Imperia, E.; Giovanetti, M.; Ceccarelli, G.; et al. Genome-based comparison between the recombinant SARS-CoV-2 XBB and its parental lineages. J. Med. Virol. 2023, 95, e28625. [Google Scholar] [CrossRef]
- Bugatti, A.; Filippini, F.; Messali, S.; Giovanetti, M.; Ravelli, C.; Zani, A.; Ciccozzi, M.; Caruso, A.; Caccuri, F. The D405N Mutation in the Spike Protein of SARS-CoV-2 Omicron BA.5 Inhibits Spike/Integrins Interaction and Viral Infection of Human Lung Microvascular Endothelial Cells. Viruses 2023, 15, 332. [Google Scholar] [CrossRef]
- Hristova, S.; Zhivkov, A. Omicron coronavirus variant: pH-dependent electrostatic potentials and energy of association of S-protein to ACE2 receptor. SSRN Electron. J. 2022, PPR605185. [Google Scholar] [CrossRef]
- Barroso da Silva, F.L.; Giron, C.C.; Laaksonen, A. Electrostatic features for the receptor binding domain of SARS-CoV-2 wildtype and its variants. Compass to the severity of the future variants with the charge-rule. bioRxiv, 2022. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Wild | Omicron |
|---|---|---|
| RBD monomer | pI 9.2 | pI 9.7 |
| S1-monomer | pI 8.7 | pI 9.2 |
| S2-monomer | pI 6.9 | pI 6.9 |
| S-monomer | pI 7.4 | pI 8.0 |
| S-trimer | pI 7.3 | pI 7.79 |
| S-trimer–ACE2 | pI 6.5 | pI 7.0 |
| Protein | Expression Region | Amino Acid Residues | Molecular Mass [kDa] | Mass Increment ΔM/M |
|---|---|---|---|---|
| ACE2 | – | 723 | 83.68 | – |
| ACE2-Fc | 18–740 | 952 | 109.4 | 31% |
| ACE2-Fc-Gly | 18–740 | 952 | 145 | 33%; 73% |
| wild RBD | 315–529 | 221 | 24.85 | – |
| wild RBD-His | 315–535 | 227 | 25.68 | 3% |
| wild RBD-His-Gly | 315–535 | 227 | 37 | 44%; 49% |
| omicron RBD | 319–535 | 223 | 25.03 | – |
| omicron RBD-His | 319–541 | 229 | 25.86 | 3% |
| omicron RBD-His-Gly | 319–541 | 229 | 33 | 28%; 32% |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hristova, S.H.; Zhivkov, A.M. Omicron Coronavirus: pH-Dependent Electrostatic Potential and Energy of Association of Spike Protein to ACE2 Receptor. Viruses 2023, 15, 1752. https://doi.org/10.3390/v15081752
Hristova SH, Zhivkov AM. Omicron Coronavirus: pH-Dependent Electrostatic Potential and Energy of Association of Spike Protein to ACE2 Receptor. Viruses. 2023; 15(8):1752. https://doi.org/10.3390/v15081752
Chicago/Turabian StyleHristova, Svetlana H., and Alexandar M. Zhivkov. 2023. "Omicron Coronavirus: pH-Dependent Electrostatic Potential and Energy of Association of Spike Protein to ACE2 Receptor" Viruses 15, no. 8: 1752. https://doi.org/10.3390/v15081752
APA StyleHristova, S. H., & Zhivkov, A. M. (2023). Omicron Coronavirus: pH-Dependent Electrostatic Potential and Energy of Association of Spike Protein to ACE2 Receptor. Viruses, 15(8), 1752. https://doi.org/10.3390/v15081752