
Evidence of a Protein-Coding Gene Antisense to the UL5 Gene in Bovine Herpesvirus I

Abstract
1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Infection
2.3. RNA Extraction
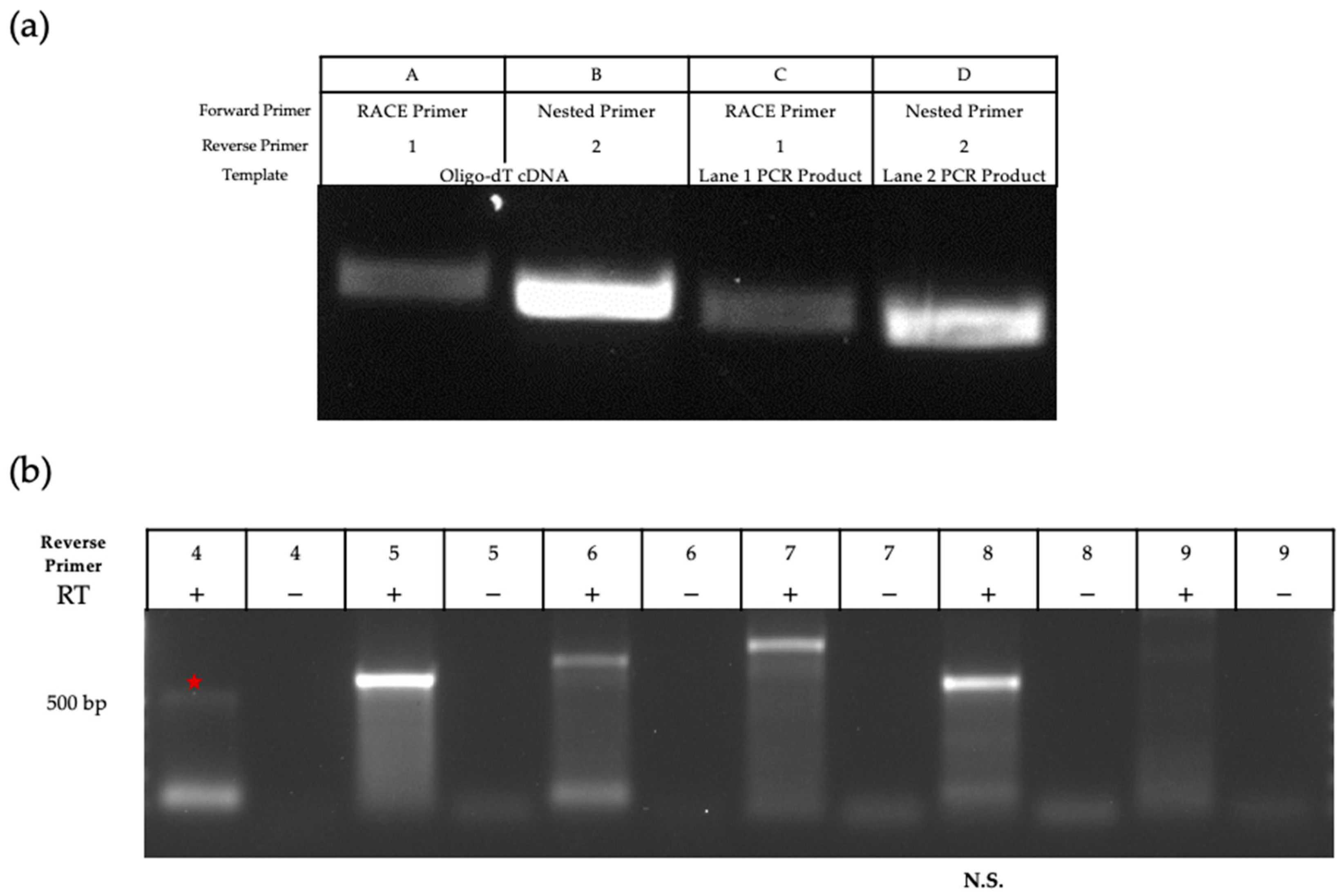
2.4. 5′ RACE, 3′ Primer Walking, Regular and Quantitative (q)RT-PCR
2.5. Immunoblot
2.6. Immunocytochemistry
2.7. Bioinformatic Analysis
3. Results
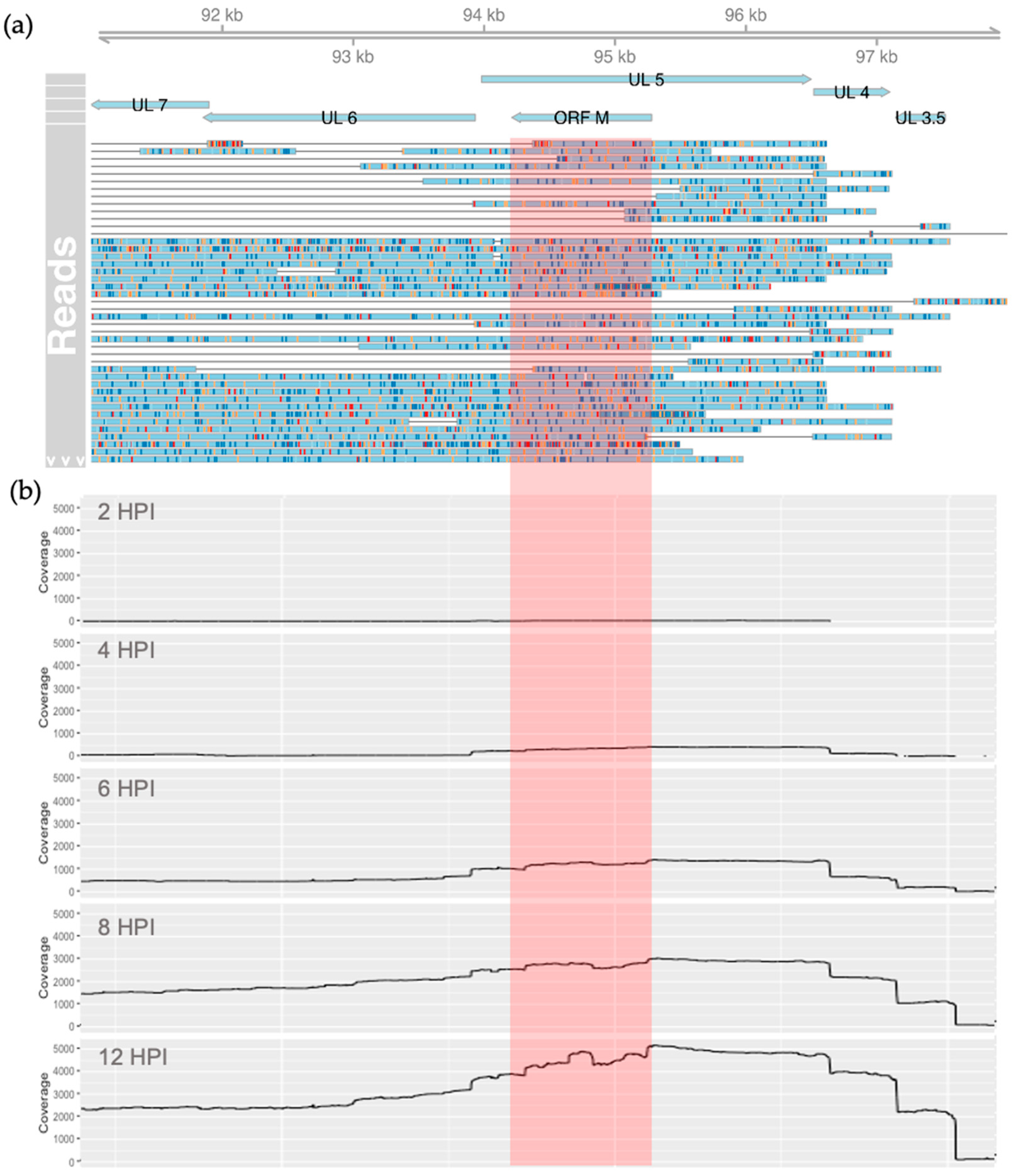
3.1. Evidence of Antisense Transcripts in the UL5 Region
3.2. Characterization of the ORF M Transcript
3.3. Detection of the ORF M Polypeptide
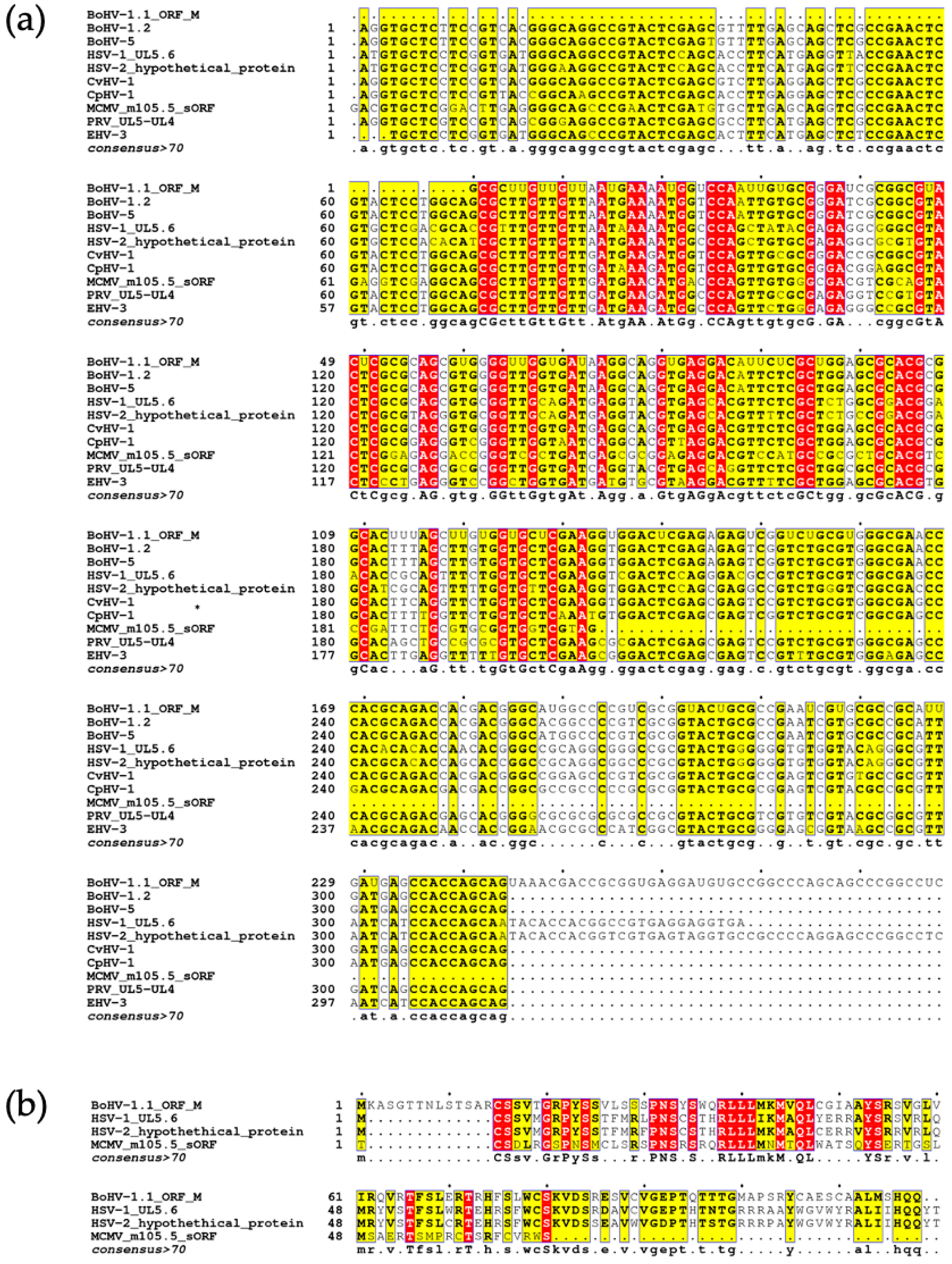
3.4. Identification of Homologous Herpesvirus Sequences
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- D’Arce, R.C.F.; Almeida, R.S.; Silva, T.C.; Franco, A.C.; Spilki, F.; Roehe, P.M.; Arns, C.W. Restriction endonuclease and monoclonal antibody analysis of Brazilian isolates of bovine herpesviruses types 1 and 5. Vet. Microbiol. 2002, 88, 315–324. [Google Scholar] [CrossRef]
- Metzler, A.E.; Matile, H.; Gassmann, U.; Engels, M.; Wyler, R. European isolates of bovine herpesvirus 1: A comparison of restriction endonuclease sites, polypeptides, and reactivity with monoclonal antibodies. Arch. Virol. 1985, 85, 57–69. [Google Scholar] [CrossRef]
- Pidone, C.L.; Galosi, C.M.; Echeverria, M.G.; Nosetto, E.O.; Etcheverrigaray, M.E. Restriction endonuclease analysis of BHV-1 and BHV-5 strains isolated in argentina. J. Vet. Med. Ser. B 1999, 46, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Thiry, J.; Keuser, V.; Muylkens, B.; Meurens, F.; Gogev, S.; Vanderplasschen, A.; Thiry, E. Ruminant alphaherpesviruses related to bovine herpesvirus 1. Vet. Res. 2006, 37, 169–190. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.; Capik, S.F.; Kegley, B.; Richeson, J.T.; Powell, J.G.; Zhao, J. Bovine respiratory microbiota of feedlot cattle and its association with disease. Vet. Res. 2022, 53, 4. [Google Scholar] [CrossRef]
- Jones, C.; Chowdhury, S. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex and development of improved vaccines. Anim. Health Res. Rev. 1996, 8, 187–205. [Google Scholar] [CrossRef]
- Kirchhoff, J.; Uhlenbruck, S.; Goris, K.; Keil, G.M.; Herrler, G. Three viruses of the bovine respiratory disease complex apply different strategies to initiate infection. Vet. Res. 2014, 45, 20. [Google Scholar] [CrossRef]
- Smith, G.A.; Young, P.L.; Reed, K.C. Emergence of a new bovine herpesvirus 1 strain in Australian feedlots. Arch. Virol. 1995, 140, 599–603. [Google Scholar] [CrossRef]
- Yates, W.D.G. A review of infectious bovine rhinotracheitis, shipping fever pneumonia and viral-bacterial synergism in respiratory disease of cattle. Can. J. Comp. Med. 1982, 46, 225. [Google Scholar]
- USDA. Morbidity Mortality in U.S. Preweaned Dairy Heifer Calves NAHMS Dairy 2014 Study Calf Component. 2021. Available online: https://www.aphis.usda.gov/animal_health/nahms/dairy/downloads/dairy17/morb-mort-us-prewean-dairy-heifer-nahms-2014.pdf (accessed on 24 May 2023).
- Enquist, L.W.; Husak, P.J.; Banfield, B.W.; Smith, G.A. Infection and spread of alphaherpesviruses in the nervous system. Adv. Virus Res. 1998, 51, 237–347. [Google Scholar] [CrossRef]
- Winkler, M.T.C.; Doster, A.; Jones, C. Persistence and Reactivation of Bovine Herpesvirus 1 in the Tonsils of Latently Infected Calves. J. Virol. 2000, 74, 5337. [Google Scholar] [CrossRef]
- Muylkens, B.; Thiry, J.; Kirten, P.; Schynts, F.; Thiry, E. Bovine herpesvirus 1 infection and infectious bovine rhinotracheitis. Vet. Res. 2007, 38, 181–209. [Google Scholar] [CrossRef] [PubMed]
- Khattar, S.K.; Van Drunen Littel-Van Den Hurk, S.; Babiuk, L.A.; Tikoo, S.K. Identification and Transcriptional Analysis of a 3′-Coterminal Gene Cluster Containing UL1, UL2, UL3, and UL3.5 Open Reading Frames of Bovine Herpesvirus-1. Virology 1995, 213, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Leung-Tack, P.; Audonnet, J.C.; Riviere, M. The Complete DNA Sequence and the Genetic Organization of the Short Unique Region (US) of the Bovine Herpesvirus Type 1 (ST Strain). Virology 1994, 199, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Simard, C.; Langlois, I.; Styger, D.; Vogt, B.; Vlcek, C.; Chalifour, A.; Trudel, M.; Schwyzer, M. Sequence analysis of the UL39, UL38, and UL37 homologues of bovine herpesvirus 1 and expression studies of UL40 and UL39, the subunits of ribonucleotide reductase. Virology 1995, 212, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Vlček, Č.; Beneš, V.; Lu, Z.; Kutish, G.F.; Pačes, V.; Rock, D.; Letchworth, G.J.; Schwyzer, M. Nucleotide Sequence Analysis of a 30-kb Region of the Bovine Herpesvirus 1 Genome Which Exhibits a Colinear Gene Arrangement with the UL21 to UL4 Genes of Herpes Simplex Virus. Virology 1995, 210, 100–108. [Google Scholar] [CrossRef]
- d’Offay, J.M.; Fulton, R.W.; Eberle, R. Complete genome sequence of the NVSL BoHV-1.1 Cooper reference strain. Arch. Virol. 2013, 158, 1109–1113. [Google Scholar] [CrossRef]
- Olson, R.D.; Assaf, R.; Brettin, T.; Conrad, N.; Cucinell, C.; Davis, J.J.; Dempsey, D.M.; Dickerman, A.; Dietrich, E.M.; Kenyon, R.W.; et al. Introducing the Bacterial and Viral Bioinformatics Resource Center (BV-BRC): A resource combining PATRIC, IRD and ViPR. Nucleic Acids Res. 2023, 51, D678–D689. [Google Scholar] [CrossRef]
- Pickett, B.; Greer, D.; Zhang, Y.; Stewart, L.; Zhou, L.; Sun, G.; Gu, Z.; Kumar, S.; Zaremba, S.; Larsen, C.; et al. Virus Pathogen Database and Analysis Resource (ViPR): A Comprehensive Bioinformatics Database and Analysis Resource for the Coronavirus Research Community. Viruses 2012, 4, 3209–3226. [Google Scholar] [CrossRef]
- Moldován, N.; Tombácz, D.; Szűcs, A.; Csabai, Z.; Snyder, M.; Boldogkői, Z. Multi-Platform Sequencing Approach Reveals a Novel Transcriptome Profile in Pseudorabies Virus. Front. Microbiol. 2018, 8, 2708. [Google Scholar] [CrossRef]
- Torma, G.; Tombácz, D.; Csabai, Z.; Göbhardter, D.; Deim, Z.; Snyder, M.; Boldogkői, Z. An Integrated Sequencing Approach for Updating the Pseudorabies Virus Transcriptome. Pathogens 2021, 10, 242. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, D.; Csabai, Z.; Oláh, P.; Havelda, Z.; Sharon, D.; Snyder, M.; Boldogkői, Z. Characterization of novel transcripts in pseudorabies virus. Viruses 2015, 7, 2727–2744. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.; Desjardins, M.; Thibault, P.; Radtke, K. Proteomics analysis of Herpes Simplex Virus type 1-infected cells reveals dynamic changes of viral protein expression, ubiquitylation, and phosphorylation. J. Proteome Res. 2013, 12, 1820–1829. [Google Scholar] [CrossRef]
- Howard, T.R.; Cristea, I.M. Interrogating Host Antiviral Environments Driven by Nuclear DNA Sensing: A Multiomic Perspective. Biomolecules 2020, 10, 1591. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Oda, S.; Watanabe, M.; Oyama, M.; Kozuka-Hata, H.; Koyanagi, N.; Maruzuru, Y.; Arii, J.; Kawaguchi, Y. Roles of the Phosphorylation of Herpes Simplex Virus 1 UL51 at a Specific Site in Viral Replication and Pathogenicity. J. Virol. 2018, 92, e01035-18. [Google Scholar] [CrossRef]
- Gatherer, D.; Seirafian, S.; Cunningham, C.; Holton, M.; Dargan, D.J.; Baluchova, K.; Hector, R.D.; Galbraith, J.; Herzyk, P.; Wilkinson, G.W.G.; et al. High-resolution human cytomegalovirus transcriptome. Proc. Natl. Acad. Sci. USA 2011, 108, 19755–19760. [Google Scholar] [CrossRef]
- Fülöp, Á.; Torma, G.; Moldován, N.; Szenthe, K.; Bánáti, F.; Almsarrhad, I.A.A.; Csabai, Z.; Tombácz, D.; Minárovits, J.; Boldogkői, Z. Integrative profiling of Epstein–Barr virus transcriptome using a multiplatform approach. Virol. J. 2022, 19, 1–17. [Google Scholar] [CrossRef]
- Moldován, N.; Torma, G.; Gulyás, G.; Hornyák, Á.; Zádori, Z.; Jefferson, V.A.; Csabai, Z.; Boldogkői, M.; Tombácz, D.; Meyer, F.; et al. Time-course profiling of bovine alphaherpesvirus 1.1 transcriptome using multiplatform sequencing. Sci. Rep. 2020, 10, 20496. [Google Scholar] [CrossRef]
- Szarka, K.; Bányai, K.; Tombácz, D.; Kakuk, B.; Torma, G.; Csabai, Z.; Gulyás, G.; Tamás, V.; Zádori, Z.; Jefferson, V.A.; et al. In-Depth Temporal Transcriptome Profiling of an Alphaherpesvirus Using Nanopore Sequencing. Viruses 2022, 14, 1289. [Google Scholar] [CrossRef]
- Tombácz, D.; Moldován, N.; Torma, G.; Nagy, T.; Hornyák, Á.; Csabai, Z.; Gulyás, G.; Boldogkői, M.; Jefferson, V.A.; Zádori, Z.; et al. Dynamic Transcriptome Sequencing of Bovine Alphaherpesvirus Type 1 and Host Cells Carried Out by a Multi-Technique Approach. Front. Genet. 2021, 12, 619056. [Google Scholar] [CrossRef]
- Jefferson, V.A.; Barber, K.A.; El-Mayet, F.S.; Jones, C.; Nanduri, B.; Meyer, F. Proteogenomic identification of a novel protein-encoding gene in bovine herpesvirus 1 that is expressed during productive infection. Viruses 2018, 10, 499. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed]
- Hahne, F.; Ivanek, R. Visualizing Genomic Data Using Gviz and Bioconductor. In Statistical Genomics: Methods and Protocols; Mathé, E., Davis, S., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 335–351. ISBN 978-1-4939-3578-9. [Google Scholar]
- Lawrence, M.; Gentleman, R.; Carey, V. rtracklayer: An R package for interfacing with genome browsers. Bioinformatics 2009, 25, 1841–1842. [Google Scholar] [CrossRef]
- Yin, T.; Cook, D.; Lawrence, M. ggbio: Visualization tools for genomic data. Genome Biol. 2012, 13, R77. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef]
- Jaffe, J.D.; Berg, H.C.; Church, G.M. Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics 2004, 4, 59–77. [Google Scholar] [CrossRef]
- Berriman, M.; Rutherford, K. Viewing and annotating sequence data with Artemis. Brief. Bioinform. 2003, 4, 124–132. [Google Scholar] [CrossRef]
- Carver, T.; Berriman, M.; Tivey, A.; Patel, C.; Böhme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.A. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [PubMed]
- Carver, T.; Harris, S.R.; Berriman, M.; Parkhill, J.; McQuillan, J.A. Artemis: An integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 2012, 28, 464–469. [Google Scholar] [CrossRef]
- Rutherford, K.; Parkhill, J.; Crook, J.; Horsnell, T.; Rice, P.; Rajandream, M.A.; Barrell, B. Artemis: Sequence visualization and annotation. Bioinformatics 2000, 16, 944–945. [Google Scholar] [CrossRef] [PubMed]
- Wirth, U.V.; Gunkel, K.; Engels, M.; Schwyzer, M. Spatial and temporal distribution of bovine herpesvirus 1 transcripts. J. Virol. 1989, 63, 4882–4889. [Google Scholar] [CrossRef] [PubMed]
- Whisnant, A.W.; Jürges, C.S.; Hennig, T.; Wyler, E.; Prusty, B.; Rutkowski, A.J.; L’hernault, A.; Djakovic, L.; Göbel, M.; Döring, K.; et al. Integrative functional genomics decodes herpes simplex virus 1. Nat. Commun. 2020, 11, 2038. [Google Scholar] [CrossRef] [PubMed]
- Lodha, M.; Muchsin, I.; Jürges, C.; Juranic Lisnic, V.; L’Hernault, A.; Rutkowski, A.J.; Prusty, B.K.; Grothey, A.; Milic, A.; Hennig, T.; et al. Decoding murine cytomegalovirus. PLOS Pathog. 2023, 19, e1010992. [Google Scholar] [CrossRef]
- Pearson, W.R. An Introduction to Sequence Similarity (“Homology”) Searching. Curr. Protoc. Bioinform. 2013, 42, 3.1.1–3.1.8. [Google Scholar] [CrossRef]
- Robinson, K.E.; Meers, J.; Gravel, J.L.; McCarthy, F.M.; Mahony, T.J. The essential and non-essential genes of Bovine herpesvirus 1. J. Gen. Virol. 2008, 89, 2851–2863. [Google Scholar] [CrossRef]
- Roychoudhury, P.; Greninger, A.L.; Jerome, K.R.; Johnston, C.; Wald, A.; Xie, H. HSV2 Whole Genome Sequences from Clinical Isolates; Laboratory Medicine, University of Washington: Seattle, WA, USA, 2018; submitted. [Google Scholar]
- Davison, A.J.; Eberle, R.; Ehlers, B.; Hayward, G.S.; McGeoch, D.J.; Minson, A.C.; Pellett, P.E.; Roizman, B.; Studdert, M.J.; Thiry, E. The order Herpesvirales. Arch. Virol. 2009, 154, 171–177. [Google Scholar] [CrossRef]
- Hay, J.; Ruyechan, W.T. Alphaherpesvirus DNA replication. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007; pp. 138–143. ISBN 978-0-511-54531-3. [Google Scholar]
- Honess, R.W. Herpes Simplex and “The Herpes Complex”: Diverse Observations and A Unifying Hypothesis The Eighth Fleming Lecture. J. Gen. Virol. 1984, 65, 2077–2107. [Google Scholar] [CrossRef]
- Haque, M.; Stanfield, B.; Kousoulas, K.G. Bovine herpesvirus type-1 glycoprotein K (gK) interacts with UL20 and is required for infectious virus production. Virology 2016, 499, 156–164. [Google Scholar] [CrossRef][Green Version]
- Lay, C.T.; Burke, J.M.; Paulsen, D.B.; Chowdhury, S.I. A Triple Gene Mutant of BoHV-1 Administered Intranasally in Lambs Replicates Efficiently in the Nasal Epithelium and Induces Neutralizing Antibody. Int. J. Vaccines Immun. 2016, 2, 1–4. [Google Scholar] [CrossRef]
- Marawan, M.A.; Deng, M.; Wang, C.; Chen, Y.; Hu, C.; Chen, J.; Chen, X.; Chen, H.; Guo, A. Characterization of BOHV-1 gG-/tk-/gE-mutant in differential protein expression, virulence, and immunity. Vet. Sci. 2021, 8, 253. [Google Scholar] [CrossRef]
- Raza, S.; Deng, M.; Shahin, F.; Yang, K.; Hu, C.; Chen, Y.; Chen, H.; Guo, A. A bovine herpesvirus 1 pUL51 deletion mutant shows impaired viral growth in vitro and reduced virulence in rabbits. Oncotarget 2016, 7, 12235–12253. [Google Scholar] [CrossRef] [PubMed]
- Beier, H.; Grimm, M. Misreading of termination codons in eukaryotes by natural nonsense suppressor tRNAs. Nucleic Acids Res. 2001, 29, 4767–4782. [Google Scholar] [CrossRef] [PubMed]
- Blanchet, S.; Cornu, D.; Argentini, M.; Namy, O. New insights into the incorporation of natural suppressor tRNAs at stop codons in Saccharomyces cerevisiae. Nucleic Acids Res. 2014, 42, 10061–10072. [Google Scholar] [CrossRef]
- Jungreis, I.; Lin, M.F.; Spokony, R.; Chan, C.S.; Negre, N.; Victorsen, A.; White, K.P.; Kellis, M. Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 2011, 21, 2096–2113. [Google Scholar] [CrossRef]
- Romero-Romero, M.L.; Kirilenko, A.; Poehls, J.; Richter, D.; Jumel, T.; Shevchenko, A.; Toth-Petroczy, A. Environment Modulates Protein Heterogeneity Through Transcriptional And Translational Stop Codon Miscoding. bioRxiv 2023. [Google Scholar] [CrossRef]
- Beznosková, P.; Bidou, L.; Namy, O.; Valášek, L.S. Increased expression of tryptophan and tyrosine tRNAs elevates stop codon readthrough of reporter systems in human cell lines. Nucleic Acids Res. 2021, 49, 5202–5215. [Google Scholar] [CrossRef]
- Jaber, T.; Yuan, Y. A Virally Encoded Small Peptide Regulates RTA Stability and Facilitates Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication. J. Virol. 2013, 87, 3461–3470. [Google Scholar] [CrossRef]
- Xu, Y.; Ganem, D. Making Sense of Antisense: Seemingly Noncoding RNAs Antisense to the Master Regulator of Kaposi’s Sarcoma-Associated Herpesvirus Lytic Replication Do Not Regulate That Transcript but Serve as mRNAs Encoding Small Peptides. J. Virol. 2010, 84, 5465. [Google Scholar] [CrossRef] [PubMed]
- Wyler, E.; Menegatti, J.; Franke, V.; Kocks, C.; Boltengagen, A.; Hennig, T.; Theil, K.; Rutkowski, A.; Ferrai, C.; Baer, L.; et al. Widespread activation of antisense transcription of the host genome during herpes simplex virus 1 infection. Genome Biol. 2017, 18, 209. [Google Scholar] [CrossRef] [PubMed]
- Erhard, F.; Halenius, A.; Zimmermann, C.; L’Hernault, A.; Kowalewski, D.J.; Weekes, M.P.; Stevanovic, S.; Zimmer, R.; Dölken, L. Improved Ribo-seq enables identification of cryptic translation events. Nat. Methods 2018, 15, 363–366. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Forward Primer | Reverse Primer |
|---|---|---|
| bGH | 5′ GCTTTCGCCCTGCTCTGCC 3′ | 5′ TCCTGCCTCCCCACCCCTA 3′ |
| bICP0 | 5′ CGTTTGTGCGCAGCCTGTTG 3′ | 5′ GACGACGACTCTTCTGACTC 3′ |
| RR | 5′ TTTTACGAGACCGAGTGCCC 3′ | 5′ GACGAAAAGGTTGTGGGTGC 3′ |
| gC | 5′ TGATCGCAGCTATTTTCGCC 3′ | 5′ TTCTGGGCTACGAACAGCAG 3′ |
| gB | 5′ CTAACATGGAGCGCCGCTT 3′ | 5′ CGGGGCGATGCCGTC 3′ |
| Histone H2A | 5′ GTCGTGGCAAGCAAGGAG 3′ | 5′ GATCTCGGCCGTTAGGTACTC 3′ |
| ORF M RT-PCR | 5′ ATGAAGGCGTCTGGCACGAC 3′ | 5′ TTACTGCTGGTGGCTCATCAATGC 3′ |
| ORF M qPCR | 5′ GTTCGCGGGGTTGTTAATCAC 3′ | 5′ CCTCACCTGCCTTATCAC 3′ |
| RACE | RACE Primer Provided by Invitrogen Nested Primer Provided by Invitrogen | 1 5′ CCATGCCCGTCGTGGTCTGCGTGGGTT 3′ 2 5′ CCCACGCAGACCGACTCTCTCGAGTCCA 3′ |
| Primer Walking | 3 5′ ATGAAGGCGTCTGGCACGAC 3′ | 4 5′ CGGGCAGTTCGAGGCGCTGC 3′ |
| 5 5′ CCAGCCCGCCCAGCATCCAG 3′ 6 5′ CCTACCACAGCCGTCACATCAACA 3′ 7 5′ CAAAAGCACCTGCATCCAAACCCTC 3′ 8 5′ CGCCGCTTGGCTGGTTCCGC 3′ 9 5′ AGCCGAGGTCTTCCTGAATTTCACT 3′ |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jefferson, V.A.; Bostick, H.; Oldenburg, D.; Meyer, F. Evidence of a Protein-Coding Gene Antisense to the UL5 Gene in Bovine Herpesvirus I. Viruses 2023, 15, 1977. https://doi.org/10.3390/v15101977
Jefferson VA, Bostick H, Oldenburg D, Meyer F. Evidence of a Protein-Coding Gene Antisense to the UL5 Gene in Bovine Herpesvirus I. Viruses. 2023; 15(10):1977. https://doi.org/10.3390/v15101977
Chicago/Turabian StyleJefferson, Victoria A., Hannah Bostick, Darby Oldenburg, and Florencia Meyer. 2023. "Evidence of a Protein-Coding Gene Antisense to the UL5 Gene in Bovine Herpesvirus I" Viruses 15, no. 10: 1977. https://doi.org/10.3390/v15101977
APA StyleJefferson, V. A., Bostick, H., Oldenburg, D., & Meyer, F. (2023). Evidence of a Protein-Coding Gene Antisense to the UL5 Gene in Bovine Herpesvirus I. Viruses, 15(10), 1977. https://doi.org/10.3390/v15101977