
Cellular Lipids—Hijacked Victims of Viruses

{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
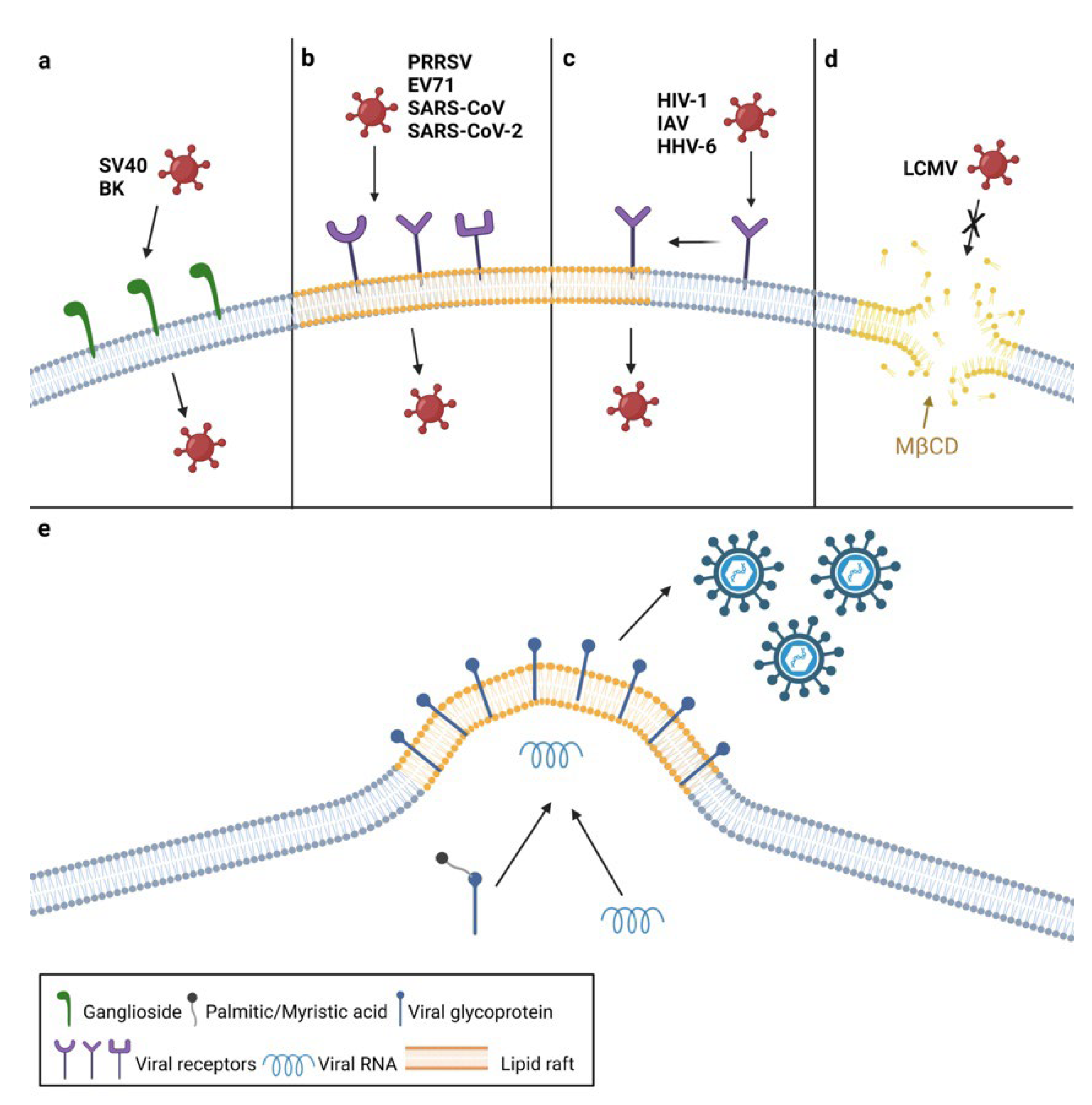
2. The Roles of Plasma Membrane Lipids in Virus Entry
3. The Roles of Plasma Membrane Lipids in Virus Assembly and Egress
4. Endosomal Membrane Lipids Facilitate Virion Delivery
5. Viruses Remodel Endoplasmic Reticulum Membranes
6. Viruses Utilize Lipids Stored in Lipid Droplets
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lorizate, M.; Kräusslich, H.G. Role of Lipids in Virus Replication. Cold Spring Harb. Perspect. Biol. 2011, 3, a004820. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Gilbert, J.M.; Stehle, T.; Lencer, W.; Benjamin, T.L.; Rapoport, T.A. Gangliosides Are Receptors for Murine Polyoma Virus and SV40. EMBO J. 2003, 22, 4346–4355. [Google Scholar] [CrossRef] [PubMed]
- Erickson, K.D.; Garcea, R.L.; Tsai, B. Ganglioside GT1b Is a Putative Host Cell Receptor for the Merkel Cell Polyomavirus. J. Virol. 2009, 83, 10275–10279. [Google Scholar] [CrossRef]
- Gilbert, J.; Benjamin, T. Uptake Pathway of Polyomavirus via Ganglioside GD1a. J. Virol. 2004, 78, 12259–12267. [Google Scholar] [CrossRef] [PubMed]
- Molina, S.; Castet, V.; Fournier-Wirth, C.; Pichard-Garcia, L.; Avner, R.; Harats, D.; Roitelman, J.; Barbaras, R.; Graber, P.; Ghersa, P.; et al. The Low-Density Lipoprotein Receptor Plays a Role in the Infection of Primary Human Hepatocytes by Hepatitis C Virus. J. Hepatol. 2007, 46, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Suhy, D.A.; Giddings, T.H.; Kirkegaard, K. Remodeling the Endoplasmic Reticulum by Poliovirus Infection and by Individual Viral Proteins: An Autophagy-Like Origin for Virus-Induced Vesicles. J. Virol. 2000, 74, 8953–8965. [Google Scholar] [CrossRef]
- Paul, D.; Bartenschlager, R. Flaviviridae Replication Organelles: Oh, What a Tangled Web We Weave. Annu. Rev. Virol. 2015, 2, 289–310. [Google Scholar] [CrossRef]
- Chen, D.; Zhao, Y.G.; Zhang, H. Endomembrane Remodeling in SARS-CoV-2 Infection. Cell Insight 2022, 1, 100031. [Google Scholar] [CrossRef]
- Romero-Brey, I.; Merz, A.; Chiramel, A.; Lee, J.Y.; Chlanda, P.; Haselman, U.; Santarella-Mellwig, R.; Habermann, A.; Hoppe, S.; Kallis, S.; et al. Three-Dimensional Architecture and Biogenesis of Membrane Structures Associated with Hepatitis C Virus Replication. PLoS Pathog. 2012, 8, e1003056. [Google Scholar] [CrossRef]
- Herker, E.; Harris, C.; Hernandez, C.; Carpentier, A.; Kaehlcke, K.; Rosenberg, A.R.; Farese, R.V.; Ott, M. Efficient Hepatitis C Virus Particle Formation Requires Diacylglycerol Acyltransferase-1. Nat. Med. 2010, 16, 1295–1298. [Google Scholar] [CrossRef]
- Levental, I.; Grzybek, M.; Simons, K. Greasing Their Way: Lipid Modifications Determine Protein Association with Membrane Rafts. Biochemistry 2010, 49, 6305–6316. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Dengue Virus-Induced Autophagy Regulates Lipid Metabolism. Cell Host Microbe 2010, 8, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Low, J.A.; Magnuson, B.; Tsai, B.; Imperiale, M.J. Identification of Gangliosides GD1b and GT1b as Receptors for BK Virus. J. Virol. 2006, 80, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Iša, P.; Realpe, M.; Romero, P.; López, S.; Arias, C.F. Rotavirus RRV Associates with Lipid Membrane Microdomains during Cell Entry. Virology 2004, 322, 370–381. [Google Scholar] [CrossRef]
- Pike, L.J. Lipid Rafts: Bringing Order to Chaos. J. Lipid Res. 2003, 44, 655–667. [Google Scholar] [CrossRef]
- Schütz, G.J.; Kada, G.; Pastushenko, V.P.; Schindler, H. Properties of Lipid Microdomains in a Muscle Cell Membrane Visualized by Single Molecule Microscopy. EMBO J. 2000, 19, 892–901. [Google Scholar] [CrossRef]
- Chazal, N.; Gerlier, D. Virus Entry, Assembly, Budding, and Membrane Rafts. Microbiol. Mol. Biol. Rev. 2003, 67, 226–237. [Google Scholar] [CrossRef]
- Ripa, I.; Andreu, S.; López-Guerrero, J.A.; Bello-Morales, R. Membrane Rafts: Portals for Viral Entry. Front. Microbiol. 2021, 12, 631274. [Google Scholar] [CrossRef]
- Yang, Q.; Zhang, Q.; Tang, J.; Feng, W.H. Lipid Rafts Both in Cellular Membrane and Viral Envelope Are Critical for PRRSV Efficient Infection. Virology 2015, 484, 170–180. [Google Scholar] [CrossRef]
- Zhu, Y.Z.; Wu, D.G.; Ren, H.; Xu, Q.Q.; Zheng, K.C.; Chen, W.; Chen, S.L.; Qian, X.J.; Tao, Q.Y.; Wang, Y.; et al. The Role of Lipid Rafts in the Early Stage of Enterovirus 71 Infection. Cell. Physiol. Biochem. 2015, 35, 1347–1359. [Google Scholar] [CrossRef]
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid Rafts Are Involved in SARS-CoV Entry into Vero E6 Cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Warner, F.J.; Lew, R.A.; Smith, A.I.; Lambert, D.W.; Hooper, N.M.; Turner, A.J. Angiotensin-Converting Enzyme 2 (ACE2), but Not ACE, Is Preferentially Localized to the Apical Surface of Polarized. J. Biol. Chem. 2005, 280, 39353–39362. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhao, R.; Gao, L.; Gao, X.; Wang, D.; Gallagher, T. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, W.; Fan, M.; Zhang, J.; Peng, Y.; Huang, F.; Wang, N.; He, L.; Zhang, L.; Holmdahl, R.; et al. Dependence of SARS-CoV-2 Infection on Cholesterol-Rich Lipid Raft and Endosomal Acidification. Comput. Struct. Biotechnol. J. 2021, 19, 1933–1943. [Google Scholar] [CrossRef]
- Kozak, S.L.; Heard, J.M.; Kabat, D. Segregation of CD4 and CXCR4 into Distinct Lipid Microdomains in T Lymphocytes Suggests a Mechanism for Membrane Destabilization by Human Immunodeficiency Virus. J. Virol. 2002, 76, 1802–1815. [Google Scholar] [CrossRef]
- Popik, W.; Alce, T.M.; Au, W.-C. Human Immunodeficiency Virus Type 1 Uses Lipid Raft-Colocalized CD4 and Chemokine Receptors for Productive Entry into CD4+ T Cells. J. Virol. 2002, 76, 4709–4722. [Google Scholar] [CrossRef]
- Mañes, S.; Del Real, G.; Lacalle, R.A.; Lucas, P.; Gómez-Moutón, C.; Sánchez-Palomino, S.; Delgado, R.; Alcamí, J.; Mira, E.; Martínez-A, C. Membrane Raft Microdomains Mediate Lateral Assemblies Required for HIV-1 Infection. EMBO Rep. 2000, 1, 190–196. [Google Scholar] [CrossRef]
- Pitha, J.; Irie, T.; Sklar, P.B.; Nye, J.S. Drug Solubilizers to Aid Pharmacologists: Amorphous Cyclodextrin Derivatives. Life Sci. 1988, 43, 493–502. [Google Scholar] [CrossRef]
- Liao, Z.; Cimakasky, L.M.; Hampton, R.; Nguyen, D.H.; Hildreth, J.E.K. Lipid Rafts and HIV Pathogenesis: Host Membrane Cholesterol Is Required for Infection by HIV Type 1. AIDS Res. Hum. Retrovir. 2001, 17, 1009–1019. [Google Scholar] [CrossRef]
- Tang, H.; Kawabata, A.; Takemoto, M.; Yamanishi, K.; Mori, Y. Human Herpesvirus-6 Infection Induces the Reorganization of Membrane Microdomains in Target Cells, Which Are Required for Virus Entry. Virology 2008, 378, 265–271. [Google Scholar] [CrossRef]
- Bender, F.C.; Whitbeck, J.C.; Ponce de Leon, M.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Specific Association of Glycoprotein B with Lipid Rafts during Herpes Simplex Virus Entry. J. Virol. 2003, 77, 9542–9552. [Google Scholar] [CrossRef] [PubMed]
- Hambleton, S.; Steinberg, S.P.; Gershon, M.D.; Gershon, A.A. Cholesterol Dependence of Varicella-Zoster Virion Entry into Target Cells. J. Virol. 2007, 81, 7548–7558. [Google Scholar] [CrossRef] [PubMed]
- Maresova, L.; Pasieka, T.J.O.; Grose, C. Varicella-Zoster Virus GB and GE Coexpression, but Not GB or GE Alone, Leads to Abundant Fusion and Syncytium Formation Equivalent to Those from GH and GL Coexpression. J. Virol. 2001, 75, 9483–9492. [Google Scholar] [CrossRef]
- Gianni, T.; Campadelli-Fiume, G. AVβ3-Integrin Relocalizes Nectin1 and Routes Herpes Simplex Virus to Lipid Rafts. J. Virol. 2012, 86, 2850–2855. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, R.P.; Geraghty, R.J. Herpes Simplex Virus Type 1 Mediates Fusion through a Hemifusion Intermediate by Sequential Activity of Glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. USA 2007, 104, 2903–2908. [Google Scholar] [CrossRef] [PubMed]
- Rothberg, K.G.; Heuser, J.E.; Donzell, W.C.; Ying, Y.S.; Glenney, J.R.; Anderson, R.G.W. Caveolin, a Protein Component of Caveolae Membrane Coats. Cell 1992, 68, 673–682. [Google Scholar] [CrossRef]
- Stang, E.; Kartenbeck, J.; Parton, R.G. Major Histocompatibility Complex Class I Molecules Mediate Association of SV40 with Caveolae. Mol. Biol. Cell 1997, 8, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Ashbourne Excoffon, K.J.D.; Moninger, T.; Zabner, J. The Coxsackie B Virus and Adenovirus Receptor Resides in a Distinct Membrane Microdomain. J. Virol. 2003, 77, 2559–2567. [Google Scholar] [CrossRef]
- Verma, D.K.; Gupta, D.; Lal, S.K. Host Lipid Rafts Play a Major Role in Binding and Endocytosis of Influenza a Virus. Viruses 2018, 10, 650. [Google Scholar] [CrossRef]
- Takeda, M.; Leser, G.P.; Russell, C.J.; Lamb, R.A. Influenza Virus Hemagglutinin Concentrates in Lipid Raft Microdomains for Efficient Viral Fusion. Proc. Natl. Acad. Sci. USA 2003, 100, 14610–14617. [Google Scholar] [CrossRef]
- Eierhoff, T.; Hrincius, E.R.; Rescher, U.; Ludwig, S.; Ehrhardt, C. The Epidermal Growth Factor Receptor (EGFR) Promotes Uptake of Influenza a Viruses (IAV) into Host Cells. PLoS Pathog. 2010, 6, e1001099. [Google Scholar] [CrossRef]
- Cao, W.; Henry, M.D.; Borrow, P.; Yamada, H.; Elder, J.H.; Ravkov, E.V.; Nichol, S.T.; Compans, R.W.; Campbell, K.P.; Oldstone, M.B.A. Identification of α-Dystroglycan as a Receptor for Lymphocytic Choriomeningitis Virus and Lassa Fever Virus. Science 1998, 282, 2079–2081. [Google Scholar] [CrossRef] [PubMed]
- Shah, W.A.; Peng, H.; Carbonetto, S. Role of Non-Raft Cholesterol in Lymphocytic Choriomeningitis Virus Infection via Alpha-Dystroglycan. J. Gen. Virol. 2006, 87, 673–678. [Google Scholar] [CrossRef] [PubMed]
- Bartenschlager, R.; Penin, F.; Lohmann, V.; André, P. Assembly of Infectious Hepatitis C Virus Particles. Trends Microbiol. 2011, 19, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Sabahi, A. Hepatitis C Virus Entry: The Early Steps in the Viral Replication Cycle. Virol. J. 2009, 6, 117. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laliberte, J.P.; McGinnes, L.W.; Peeples, M.E.; Morrison, T.G. Integrity of Membrane Lipid Rafts Is Necessary for the Ordered Assembly and Release of Infectious Newcastle Disease Virus Particles. J. Virol. 2006, 80, 10652–10662. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laliberte, J.P.; McGinnes, L.W.; Morrison, T.G. Incorporation of Functional HN-F Glycoprotein-Containing Complexes into Newcastle Disease Virus Is Dependent on Cholesterol and Membrane Lipid Raft Integrity. J. Virol. 2007, 81, 10636–10648. [Google Scholar] [CrossRef] [PubMed]
- McDonald, T.P.; Pitt, A.R.; Brown, G.; Rixon, H.W.M.L.; Sugrue, R.J. Evidence that the Respiratory Syncytial Virus Polymerase Complex Associates with Lipid Rafts in Virus-Infected Cells: A Proteomic Analysis. Virology 2004, 330, 147–157. [Google Scholar] [CrossRef]
- Vincent, S.; Gerlier, D.; Manié, S.N. Measles Virus Assembly within Membrane Rafts. J. Virol. 2000, 74, 9911–9915. [Google Scholar] [CrossRef]
- Ali, A.; Nayak, D.P. Assembly of Sendal Virus: M Protein Interacts with F and HN Proteins and with the Cytoplasmic Tail and Transmembrane Domain of F Protein. Virology 2000, 276, 289–303. [Google Scholar] [CrossRef]
- Zhang, J.; Pekosz, A.; Lamb, R.A. Influenza Virus Assembly and Lipid Raft Microdomains: A Role for the Cytoplasmic Tails of the Spike Glycoproteins. J. Virol. 2000, 74, 4634–4644. [Google Scholar] [CrossRef] [PubMed]
- Melkonian, K.A.; Ostermeyer, A.G.; Chen, J.Z.; Roth, M.G.; Brown, D.A. Role of Lipid Modifications in Targeting Proteins to Detergent-Resistant Membrane Rafts. Many Raft Proteins Are Acylated, While Few Are Prenylated. J. Biol. Chem. 1999, 274, 3910–3917. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.H.; Hildreth, J.E.K. Evidence for Budding of Human Immunodeficiency Virus Type 1 Selectively from Glycolipid-Enriched Membrane Lipid Rafts. J. Virol. 2000, 74, 3264–3272. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Reitz, M.S.; Tschachler, E.; Gallo, R.C.; Sarngadharan, M.G.; di Marzo Veronese, F. Myristoylation of Gag Proteins of HIV-1 Plays an Important Role in Virus Assembly. AIDS Res. Hum. Retrovir. 1990, 6, 721–730. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.H.; Plemenitas, A.; Fielding, C.J.; Peterlin, B.M. Nef Increases the Synthesis of and Transports Cholesterol to Lipid Rafts and HIV-1 Progeny Virions. Proc. Natl. Acad. Sci. USA 2003, 100, 8460–8465. [Google Scholar] [CrossRef]
- Ono, A.; Ablan, S.D.; Lockett, S.J.; Nagashima, K.; Freed, E.O. Phosphatidylinositol (4,5) Bisphosphate Regulates HIV-1 Gag Targeting to the Plasma Membrane. Proc. Natl. Acad. Sci. USA 2004, 101, 14889–14894. [Google Scholar] [CrossRef]
- Chukkapalli, V.; Hogue, I.B.; Boyko, V.; Hu, W.-S.; Ono, A. Interaction between the Human Immunodeficiency Virus Type 1 Gag Matrix Domain and Phosphatidylinositol-(4,5)-Bisphosphate Is Essential for Efficient Gag Membrane Binding. J. Virol. 2008, 82, 2405–2417. [Google Scholar] [CrossRef]
- Chen, B.K. T Cell Virological Synapses and HIV-1 Pathogenesis. Immunol. Res. 2012, 54, 133–139. [Google Scholar] [CrossRef]
- Jolly, C.; Sattentau, Q.J. Human Immunodeficiency Virus Type 1 Virological Synapse Formation in T Cells Requires Lipid Raft Integrity. J. Virol. 2005, 79, 12088–12094. [Google Scholar] [CrossRef]
- Nour, A.M.; Modis, Y. Endosomal Vesicles as Vehicles for Viral Genomes. Trends Cell Biol. 2014, 24, 449–454. [Google Scholar] [CrossRef]
- Kielian, M.; Klimjack, M.R.; Ghosh, S.; Duffus, W.A. Mechanisms of Mutations Inhibiting Fusion and Infection by Semliki Forest Virus. J. Cell Biol. 1996, 134, 863–872. [Google Scholar] [CrossRef] [PubMed]
- Ahn, A.; Gibbons, D.L.; Kielian, M. The Fusion Peptide of Semliki Forest Virus Associates with Sterol-Rich Membrane Domains. J. Virol. 2002, 76, 3267–3275. [Google Scholar] [CrossRef] [PubMed]
- Hoornweg, T.E.; van Duijl-Richter, M.K.S.; Ayala Nuñez, N.V.; Albulescu, I.C.; van Hemert, M.J.; Smit, J.M. Dynamics of Chikungunya Virus Cell Entry Unraveled by Single-Virus Tracking in Living Cells. J. Virol. 2016, 90, 4745–4756. [Google Scholar] [CrossRef] [PubMed]
- Moesker, B.; Rodenhuis-Zybert, I.A.; Meijerhof, T.; Wilschut, J.; Smit, J.M. Characterization of the Functional Requirements of West Nile Virus Membrane Fusion. J. Gen. Virol. 2010, 91, 389–393. [Google Scholar] [CrossRef]
- Kobayashi, T.; Hirabayashi, Y. Lipid Membrane Domains in Cell Surface and Vacuolar Systems. Glycoconj. J. 2000, 17, 163–171. [Google Scholar] [CrossRef]
- Hullin-Matsuda, F.; Luquain-Costaz, C.; Bouvier, J.; Delton-Vandenbroucke, I. Bis(Monoacylglycero)Phosphate, a Peculiar Phospholipid to Control the Fate of Cholesterol: Implications in Pathology. Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 313–324. [Google Scholar] [CrossRef]
- Roth, S.L.; Whittaker, G.R. Promotion of Vesicular Stomatitis Virus Fusion by the Endosome-Specific Phospholipid Bis(Monoacylglycero)Phosphate (BMP). FEBS Lett. 2011, 585, 865–869. [Google Scholar] [CrossRef]
- Zaitseva, E.; Yang, S.T.; Melikov, K.; Pourmal, S.; Chernomordik, L.V. Dengue Virus Ensures Its Fusion in Late Endosomes Using Compartment-Specific Lipids. PLoS Pathog. 2010, 6, e1001131. [Google Scholar] [CrossRef]
- Pattanakitsakul, S.N.; Poungsawai, J.; Kanlaya, R.; Sinchaikul, S.; Chen, S.T.; Thongboonkerd, V. Association of Alix with Late Endosomal Lysobisphosphatidic Acid Is Important for Dengue Virus Infection in Human Endothelial Cells. J. Proteome Res. 2010, 9, 4640–4648. [Google Scholar] [CrossRef]
- Pasqual, G.; Rojek, J.M.; Masin, M.; Chatton, J.Y.; Kunz, S. Old World Arenaviruses Enter the Host Cell via the Multivesicular Body and Depend on the Endosomal Sorting Complex Required for Transport. PLoS Pathog. 2011, 7, e1002232. [Google Scholar] [CrossRef]
- Markosyan, R.M.; Marin, M.; Zhang, Y.; Cohen, F.S.; Melikyan, G.B. The Late Endosome-Resident Lipid Bis(Monoacylglycero)Phosphate Is a Cofactor for Lassa Virus Fusion. PLoS Pathog. 2021, 17, e1009488. [Google Scholar] [CrossRef] [PubMed]
- Carrière, F.; Longhi, S.; Record, M. The Endosomal Lipid Bis(Monoacylglycero) Phosphate as a Potential Key Player in the Mechanism of Action of Chloroquine against SARS-CoV-2 and Other Enveloped Viruses Hijacking the Endocytic Pathway. Biochimie 2020, 179, 237–246. [Google Scholar] [CrossRef] [PubMed]
- Luquain-Costaz, C.; Rabia, M.; Hullin-Matsuda, F.; Delton, I. Bis(Monoacylglycero)Phosphate, an Important Actor in the Host Endocytic Machinery Hijacked by SARS-CoV-2 and Related Viruses. Biochimie 2020, 179, 247–256. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Bartenschlager, R. Endoplasmic Reticulum: The Favorite Intracellular Niche for Viral Replication and Assembly. Viruses 2016, 8, 160. [Google Scholar] [CrossRef] [PubMed]
- Heaton, N.S.; Randall, G. Multifaceted Roles for Lipids in Viral Infection. Trends Microbiol. 2011, 19, 368–375. [Google Scholar] [CrossRef]
- Welsch, S.; Miller, S.; Romero-Brey, I.; Merz, A.; Bleck, C.K.E.; Walther, P.; Fuller, S.D.; Antony, C.; Krijnse-Locker, J.; Bartenschlager, R. Composition and Three-Dimensional Architecture of the Dengue Virus Replication and Assembly Sites. Cell Host Microbe 2009, 5, 365–375. [Google Scholar] [CrossRef]
- Gillespie, L.K.; Hoenen, A.; Morgan, G.; Mackenzie, J.M. The Endoplasmic Reticulum Provides the Membrane Platform for Biogenesis of the Flavivirus Replication Complex. J. Virol. 2010, 84, 10438–10447. [Google Scholar] [CrossRef]
- Cerikan, B.; Goellner, S.; Neufeldt, C.J.; Haselmann, U.; Mulder, K.; Chatel-Chaix, L.; Cortese, M.; Bartenschlager, R. A Non-Replicative Role of the 3′ Terminal Sequence of the Dengue Virus Genome in Membranous Replication Organelle Formation. Cell Rep. 2020, 32, 107859. [Google Scholar] [CrossRef]
- Junjhon, J.; Pennington, J.G.; Edwards, T.J.; Perera, R.; Lanman, J.; Kuhn, R.J. Ultrastructural Characterization and Three-Dimensional Architecture of Replication Sites in Dengue Virus-Infected Mosquito Cells. J. Virol. 2014, 88, 4687–4697. [Google Scholar] [CrossRef]
- Överby, A.K.; Popov, V.L.; Niedrig, M.; Weber, F. Tick-Borne Encephalitis Virus Delays Interferon Induction and Hides Its Double-Stranded RNA in Intracellular Membrane Vesicles. J. Virol. 2010, 84, 8470–8483. [Google Scholar] [CrossRef]
- Miorin, L.; Romero-Brey, I.; Maiuri, P.; Hoppe, S.; Krijnse-Locker, J.; Bartenschlager, R.; Marcello, A. Three-Dimensional Architecture of Tick-Borne Encephalitis Virus Replication Sites and Trafficking of the Replicated RNA. J. Virol. 2013, 87, 6469–6481. [Google Scholar] [CrossRef] [PubMed]
- Gosert, R.; Egger, D.; Lohmann, V.; Bartenschlager, R.; Blum, H.E.; Bienz, K.; Moradpour, D. Identification of the Hepatitis C Virus RNA Replication Complex in Huh-7 Cells Harboring Subgenomic Replicons. J. Virol. 2003, 77, 5487–5492. [Google Scholar] [CrossRef] [PubMed]
- Romero-Brey, I.; Berger, C.; Kallis, S.; Kolovou, A.; Paul, D.; Lohmann, V.; Bartenschlager, R. NS5A Domain 1 and Polyprotein Cleavage Kinetics Are Critical for Induction of Double-Membrane Vesicles Associated with Hepatitis c Virus Replication. MBio 2015, 6, e00759-15. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Hijikata, M.; Yamaji, M.; Hosaka, M.; Takahashi, H.; Shimotohno, K. Hepatitis C Virus Non-Structural Proteins in the Probable Membranous Compartment Function in Viral Genome Replication. J. Biol. Chem. 2003, 278, 50301–50308. [Google Scholar] [CrossRef]
- Quinkert, D.; Bartenschlager, R.; Lohmann, V. Quantitative Analysis of the Hepatitis C Virus Replication Complex. J. Virol. 2005, 79, 13594–13605. [Google Scholar] [CrossRef]
- Paul, D.; Hoppe, S.; Saher, G.; Krijnse-Locker, J.; Bartenschlager, R. Morphological and Biochemical Characterization of the Membranous Hepatitis C Virus Replication Compartment. J. Virol. 2013, 87, 10612–10627. [Google Scholar] [CrossRef]
- Yu, G.-Y.; Lee, K.-J.; Gao, L.; Lai, M.M.C. Palmitoylation and Polymerization of Hepatitis C Virus NS4B Protein. J. Virol. 2006, 80, 6013–6023. [Google Scholar] [CrossRef]
- Ziebuhr, J.; Snijder, E.J.; Gorbalenya, A.E. Virus-Encoded Proteinases and Proteolytic Processing in the Nidovirales. J. Gen. Virol. 2000, 81, 853–879. [Google Scholar] [CrossRef]
- Gosert, R.; Kanjanahaluethai, A.; Egger, D.; Bienz, K.; Baker, S.C. RNA Replication of Mouse Hepatitis Virus Takes Place at Double-Membrane Vesicles. J. Virol. 2002, 76, 3697–3708. [Google Scholar] [CrossRef]
- de Wilde, A.H.; Raj, V.S.; Oudshoorn, D.; Bestebroer, T.M.; van Nieuwkoop, S.; Limpens, R.W.A.L.; Posthuma, C.C.; van der Meer, Y.; Bárcena, M.; Haagmans, B.L.; et al. MERS-Coronavirus Replication Induces Severe in Vitro Cytopathology and Is Strongly Inhibited by Cyclosporin A or Interferon-α Treatment. J. Gen. Virol. 2013, 94, 1749–1760. [Google Scholar] [CrossRef]
- Cortese, M.; Goellner, S.; Acosta, E.G.; Neufeldt, C.J.; Oleksiuk, O.; Lampe, M.; Haselmann, U.; Funaya, C.; Schieber, N.; Ronchi, P.; et al. Ultrastructural Characterization of Zika Virus Replication Factories. Cell Rep. 2017, 18, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- Knoops, K.; Kikkert, M.; Van Den Worm, S.H.E.; Zevenhoven-Dobbe, J.C.; Van Der Meer, Y.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-Coronavirus Replication Is Supported by a Reticulovesicular Network of Modified Endoplasmic Reticulum. PLoS Biol. 2008, 6, 1957–1974. [Google Scholar] [CrossRef]
- Ulasli, M.; Verheije, M.H.; de Haan, C.A.M.; Reggiori, F. Qualitative and Quantitative Ultrastructural Analysis of the Membrane Rearrangements Induced by Coronavirus. Cell. Microbiol. 2010, 12, 844–861. [Google Scholar] [CrossRef]
- Kory, N.; Farese, R.V.; Walther, T.C. Targeting Fat: Mechanisms of Protein Localization to Lipid Droplets. Trends Cell Biol. 2016, 26, 535–546. [Google Scholar] [CrossRef]
- Farese, R.V.; Walther, T.C. Lipid Droplets Finally Get a Little R-E-S-P-E-C-T. Cell 2009, 139, 855–860. [Google Scholar] [CrossRef] [PubMed]
- Filipe, A.; McLauchlan, J. Hepatitis C Virus and Lipid Droplets: Finding a Niche. Trends Mol. Med. 2015, 21, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, K.M.; Binns, D.; Bartz, R.; Grishin, N.V.; Li, W.P.; Agarwal, A.K.; Garg, A.; Anderson, R.G.W.; Goodman, J.M. The Lipodystrophy Protein Seipin Is Found at Endoplasmic Reticulum Lipid Droplet Junctions and Is Important for Droplet Morphology. Proc. Natl. Acad. Sci. USA 2007, 104, 20890–20895. [Google Scholar] [CrossRef]
- Wu, H.; Carvalho, P.; Voeltz, G.K. Here, There, and Everywhere: The Importance of ER Membrane Contact Sites. Science 2018, 361, eaan5835. [Google Scholar] [CrossRef]
- Herker, E.; Ott, M. Emerging Role of Lipid Droplets in Host/Pathogen Interactions. J. Biol. Chem. 2012, 287, 2280–2287. [Google Scholar] [CrossRef]
- Appel, N.; Zayas, M.; Miller, S.; Krijnse-Locker, J.; Schaller, T.; Friebe, P.; Kallis, S.; Engel, U.; Bartenschlager, R. Essential Role of Domain III of Nonstructural Protein 5A for Hepatitis C Virus Infectious Particle Assembly. PLoS Pathog. 2008, 4, e1000035. [Google Scholar] [CrossRef]
- Rouillé, Y.; Helle, F.; Delgrange, D.; Roingeard, P.; Voisset, C.; Blanchard, E.; Belouzard, S.; McKeating, J.; Patel, A.H.; Maertens, G.; et al. Subcellular Localization of Hepatitis C Virus Structural Proteins in a Cell Culture System that Efficiently Replicates the Virus. J. Virol. 2006, 80, 2832–2841. [Google Scholar] [CrossRef] [PubMed]
- Miyanari, Y.; Atsuzawa, K.; Usuda, N.; Watashi, K.; Hishiki, T.; Zayas, M.; Bartenschlager, R.; Wakita, T.; Hijikata, M.; Shimotohno, K. The Lipid Droplet Is an Important Organelle for Hepatitis C Virus Production. Nat. Cell Biol. 2007, 9, 1089–1097. [Google Scholar] [CrossRef] [PubMed]
- Majeau, N.; Frometin, R.; Savard, C.; Duval, M.; Tremblay, M.J.; Leclerc, D. Palmitoylation of Hepatitis C Virus Core Protein Is Important for Virion Production. J. Biol. Chem. 2009, 284, 33915–33925. [Google Scholar] [CrossRef] [PubMed]
- Eberlé, D.; Hegarty, B.; Bossard, P.; Ferré, P.; Foufelle, F. SREBP Transcription Factors: Master Regulators of Lipid Homeostasis. Biochimie 2004, 86, 839–848. [Google Scholar] [CrossRef] [PubMed]
- Huang, H.; Sun, F.; Owen, D.M.; Li, W.; Chen, Y.; Gale, M.; Ye, J. Hepatitis C Virus Production by Human Hepatocytes Dependent on Assembly and Secretion of Very Low-Density Lipoproteins. Proc. Natl. Acad. Sci. USA 2007, 104, 5848–5853. [Google Scholar] [CrossRef]
- Gastaminza, P.; Dryden, K.A.; Boyd, B.; Wood, M.R.; Law, M.; Yeager, M.; Chisari, F.V. Ultrastructural and Biophysical Characterization of Hepatitis C Virus Particles Produced in Cell Culture. J. Virol. 2010, 84, 10999–11009. [Google Scholar] [CrossRef]
- Chang, K.-S.; Jiang, J.; Cai, Z.; Luo, G. Human Apolipoprotein E Is Required for Infectivity and Production of Hepatitis C Virus in Cell Culture. J. Virol. 2007, 81, 13783–13793. [Google Scholar] [CrossRef]
- Meunier, J.-C.; Russell, R.S.; Goossens, V.; Priem, S.; Walter, H.; Depla, E.; Union, A.; Faulk, K.N.; Bukh, J.; Emerson, S.U.; et al. Isolation and Characterization of Broadly Neutralizing Human Monoclonal Antibodies to the E1 Glycoprotein of Hepatitis C Virus. J. Virol. 2008, 82, 966–973. [Google Scholar] [CrossRef]
- Long, G.; Hiet, M.; Windisch, M.P.; Lee, J.; Lohmann, V.; Bartenschlager, R. Mouse Hepatic Cells Support Assembly of Infectious Hepatitis C Virus Particles. Gastroenterology 2011, 141, 1057–1066. [Google Scholar] [CrossRef]
- Cheung, W.; Gill, M.; Esposito, A.; Kaminski, C.F.; Courousse, N.; Chwetzoff, S.; Trugnan, G.; Keshavan, N.; Lever, A.; Desselberger, U. Rotaviruses Associate with Cellular Lipid Droplet Components To Replicate in Viroplasms, and Compounds Disrupting or Blocking Lipid Droplets Inhibit Viroplasm Formation and Viral Replication. J. Virol. 2010, 84, 6782–6798. [Google Scholar] [CrossRef]
- Criglar, J.M.; Estes, M.K.; Crawford, S.E. Rotavirus-Induced Lipid Droplet Biogenesis Is Critical for Virus Replication. Front. Physiol. 2022, 13, 836870. [Google Scholar] [CrossRef] [PubMed]
- da Silva Gomes Dias, S.; Soares, V.C.; Ferreira, A.C.; Sacramento, C.Q.; Fintelman-Rodrigues, N.; Temerozo, J.R.; Teixeira, L.; da Silva, M.A.N.; Barreto, E.; Mattos, M.; et al. Lipid Droplets Fuel SARS-CoV-2 Replication and Production of Inflammatory Mediators. PLoS Pathog. 2020, 16, e1009127. [Google Scholar]
- Ricciardi, S.; Guarino, A.M.; Giaquinto, L.; Polishchuk, E.V.; Santoro, M.; Di Tullio, G.; Wilson, C.; Panariello, F.; Soares, V.C.; Dias, S.S.G.; et al. The Role of NSP6 in the Biogenesis of the SARS-CoV-2 Replication Organelle. Nature 2022, 606, 761–768. [Google Scholar] [CrossRef] [PubMed]
- Kudchodkar, S.B.; Levine, B. Viruses and Autophagy. Rev. Med. Virol. 2009, 19, 359–378. [Google Scholar] [CrossRef]
- Zhang, J.; Lan, Y.; Li, M.Y.; Lamers, M.M.; Fusade-Boyer, M.; Klemm, E.; Thiele, C.; Ashour, J.; Sanyal, S. Flaviviruses Exploit the Lipid Droplet Protein AUP1 to Trigger Lipophagy and Drive Virus Production. Cell Host Microbe 2018, 23, 819–831.e5. [Google Scholar] [CrossRef]
- Sahni, S.; Bae, D.H.; Lane, D.J.R.; Kovacevic, Z.; Kalinowski, D.S.; Jansson, P.J.; Richardson, D.R. The Metastasis Suppressor, N-Myc Downstream-Regulated Gene 1 (NDRG1), Inhibits Stress-Induced Autophagy in Cancer Cells. J. Biol. Chem. 2014, 289, 9692–9709. [Google Scholar] [CrossRef]
- Wang, J.; Liu, J.-Y.; Shao, K.-Y.; Han, Y.-Q.; Li, G.-L.; Ming, S.-L.; Su, B.-Q.; Du, Y.-K.; Liu, Z.-H.; Zhang, G.-P.; et al. Porcine Reproductive and Respiratory Syndrome Virus Activates Lipophagy To Facilitate Viral Replication through Downregulation of NDRG1 Expression. J. Virol. 2019, 93, e00526-19. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Omasta, B.; Tomaskova, J. Cellular Lipids—Hijacked Victims of Viruses. Viruses 2022, 14, 1896. https://doi.org/10.3390/v14091896
Omasta B, Tomaskova J. Cellular Lipids—Hijacked Victims of Viruses. Viruses. 2022; 14(9):1896. https://doi.org/10.3390/v14091896
Chicago/Turabian StyleOmasta, Bozena, and Jana Tomaskova. 2022. "Cellular Lipids—Hijacked Victims of Viruses" Viruses 14, no. 9: 1896. https://doi.org/10.3390/v14091896
APA StyleOmasta, B., & Tomaskova, J. (2022). Cellular Lipids—Hijacked Victims of Viruses. Viruses, 14(9), 1896. https://doi.org/10.3390/v14091896