

Detection and Characterization of a Reassortant Mammalian Orthoreovirus Isolated from Bats in Xinjiang, China
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Information and Virus Detection
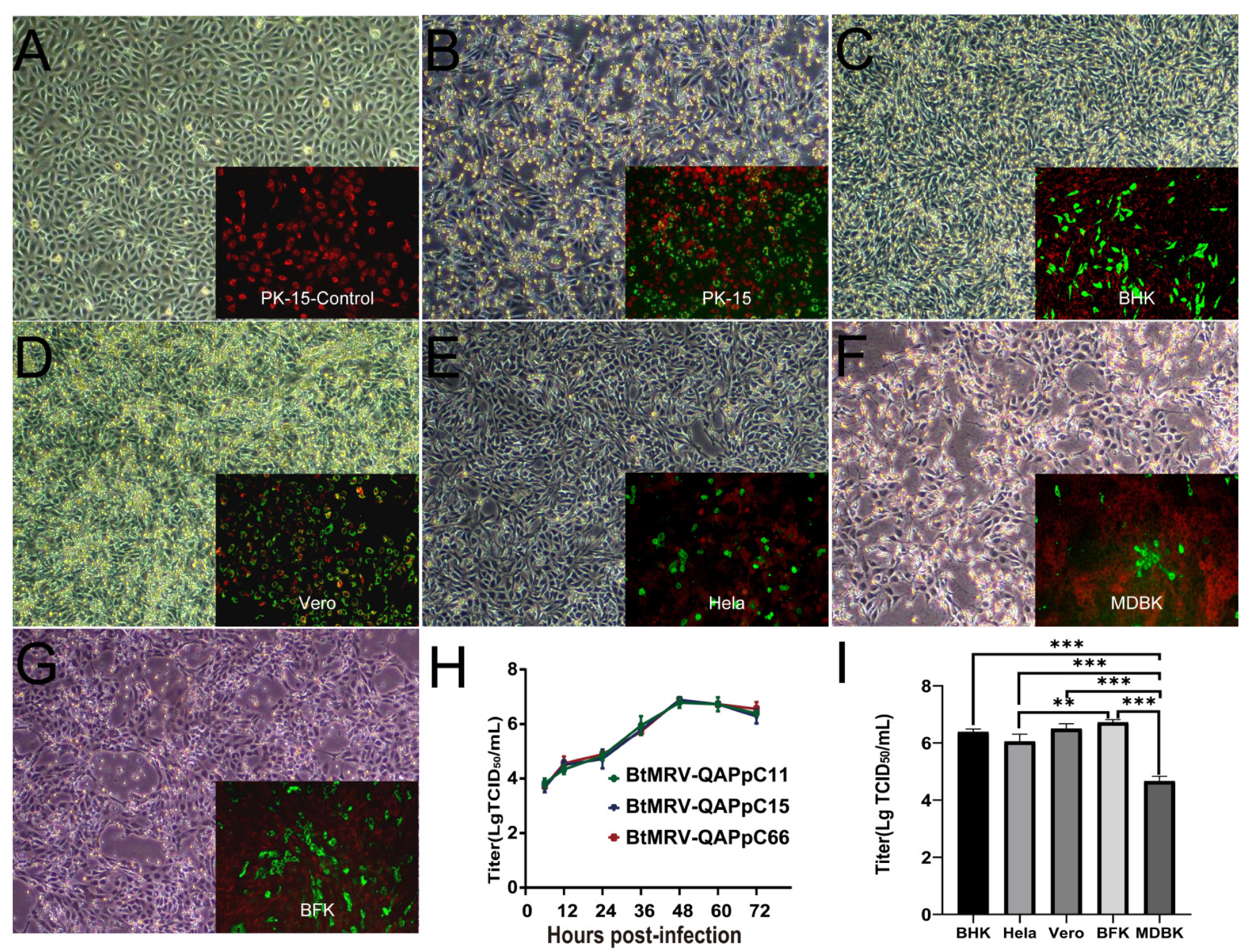
2.2. Virus Isolation and Cell Tropism Test
2.3. Genome Sequencing and Phylogenetic Analysis
2.4. Reassortment Analysis
2.5. Experimental Infection
2.6. Statistical Analysis
3. Results
3.1. Detection of MRV RNA in Bats
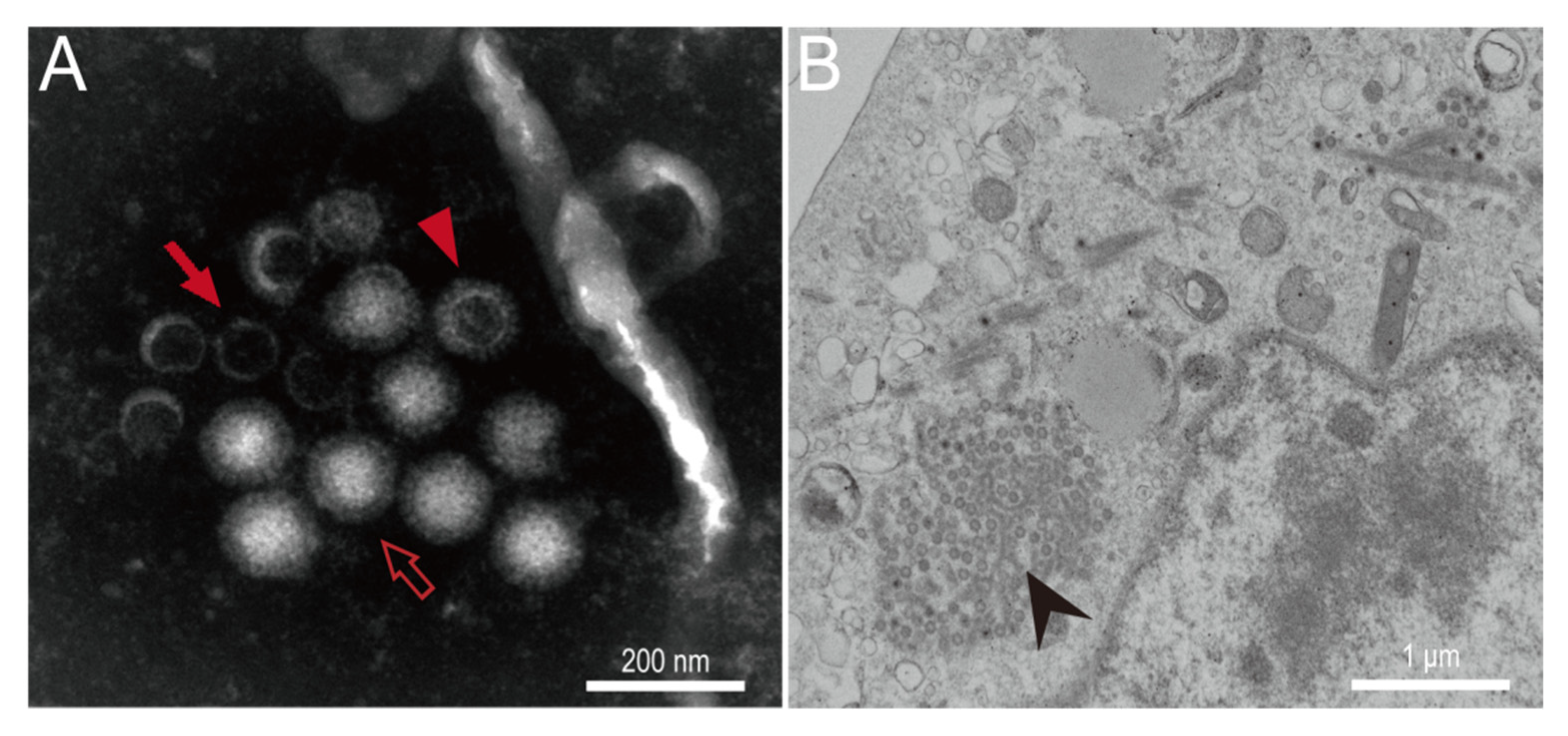
3.2. In Vitro Isolation and Characterization of MRV
3.3. Results of Experimental Infection
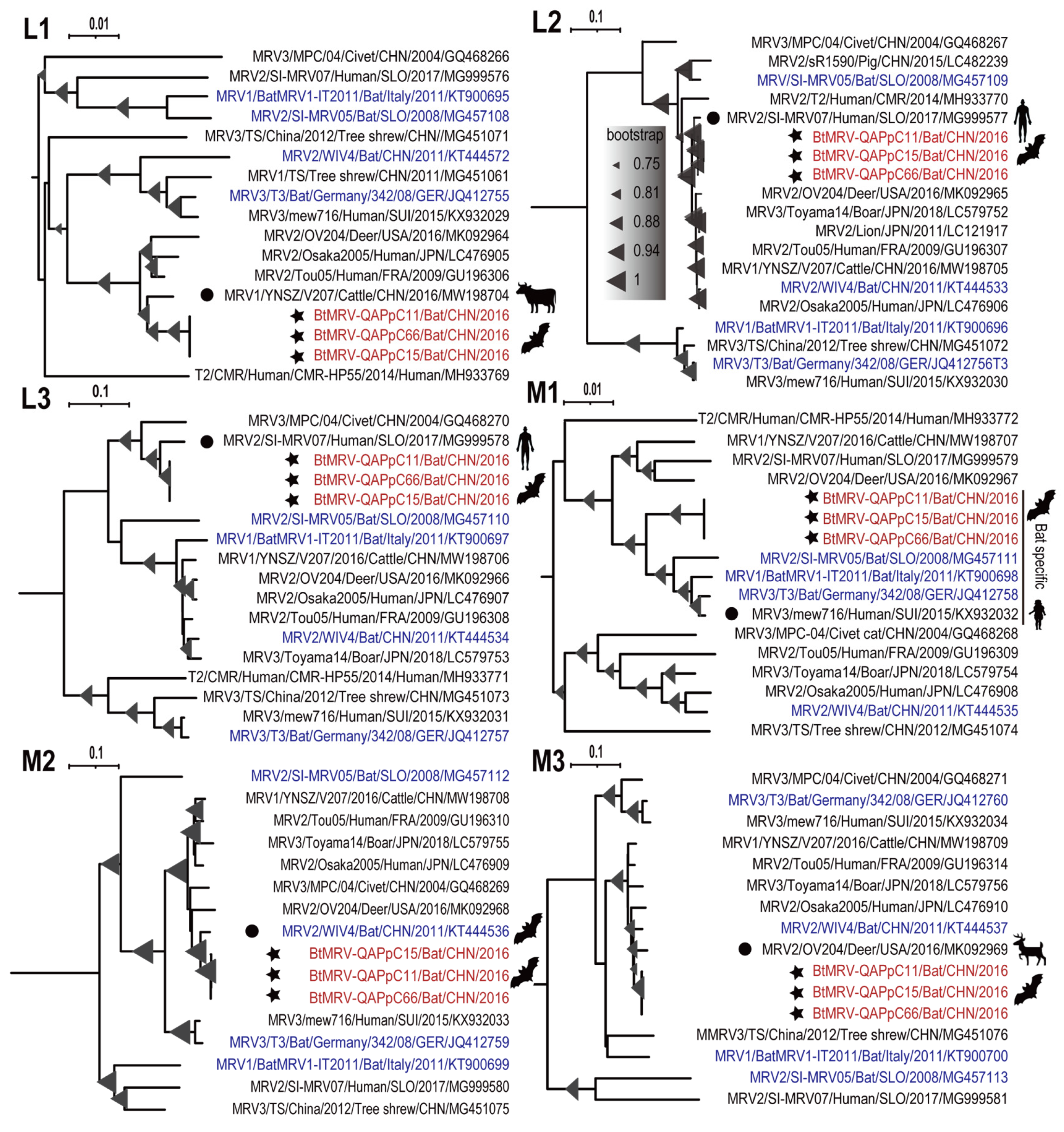
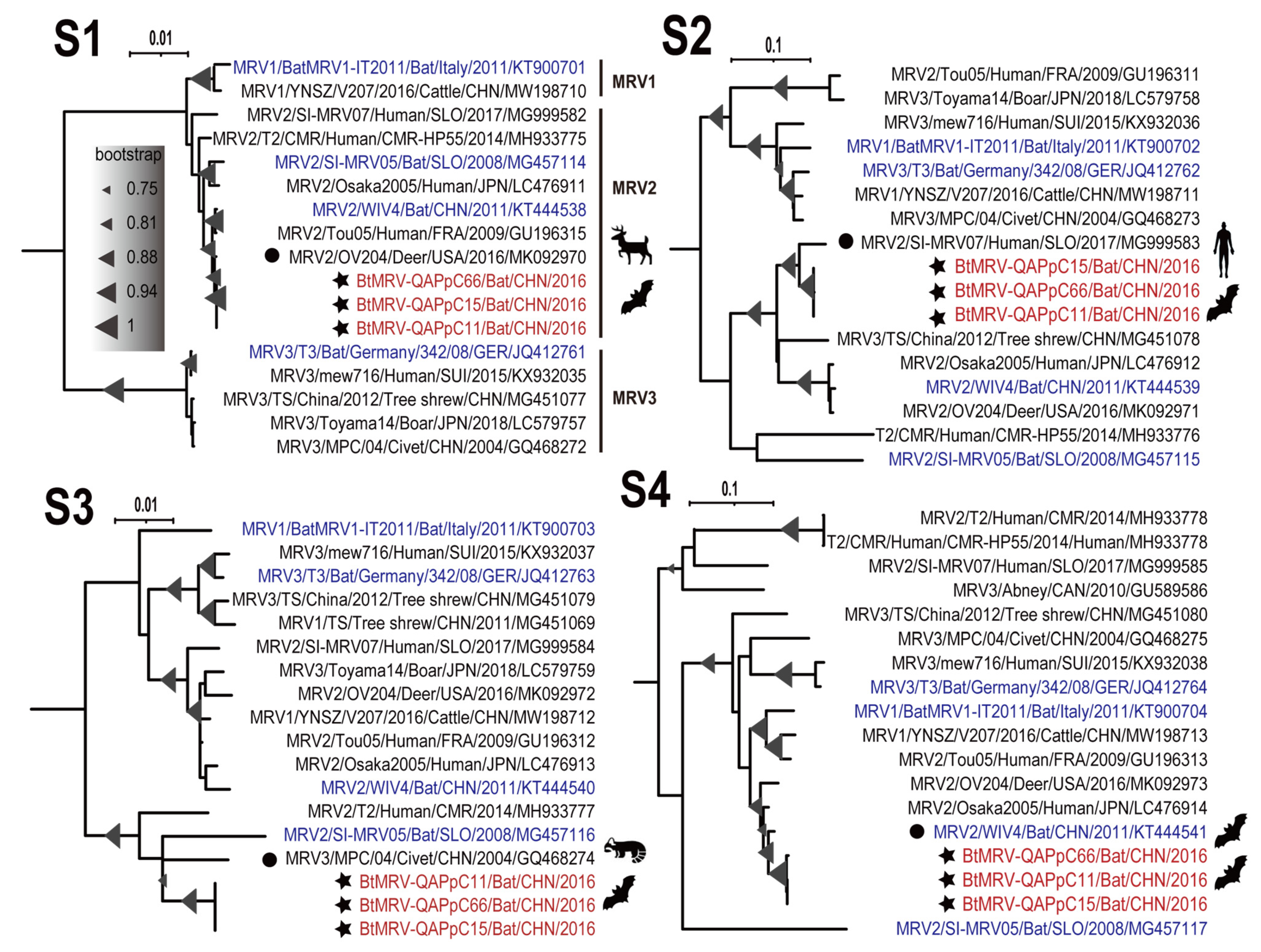
3.4. Full Genome Characterization and Phylogenetic Analyses
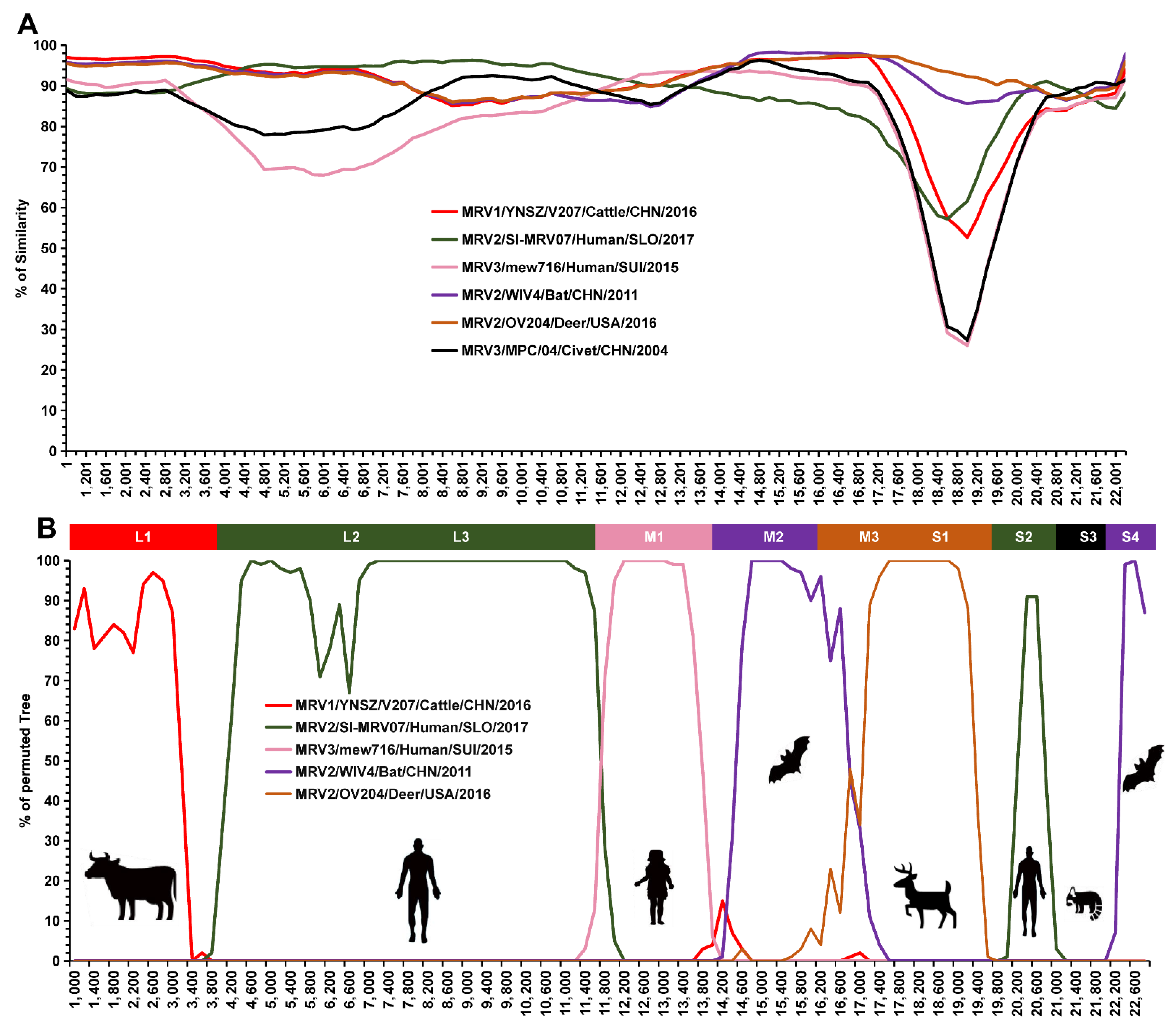
3.5. Reassortment Analysis
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Matthijnssens, J.; Attoui, H.; Bányai, K.; Brussaard, C.P.D.; Danthi, P.; del Vas, M.; Dermody, T.S.; Duncan, R.; Fāng, Q.; Johne, R.; et al. ICTV Virus Taxonmy Profile: Sedoreoviridae J. Gen. Virol. 2022, in press. [Google Scholar]
- Coombs, K.M. Reovirus structure and morphogenesis. Curr. Top. Microbiol. Immunol. 2006, 309, 117–167. [Google Scholar] [PubMed]
- Xu, W.; Coombs, K.M. Conserved structure/function of the orthoreovirus major core proteins. Virus Res. 2009, 144, 44–57. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kumar, S.; Dick, E.J., Jr.; Bommineni, Y.R.; Yang, A.; Mubiru, J.; Hubbard, G.B.; Owston, M.A. Reovirus-associated meningoencephalomyelitis in baboons. Vet. Pathol. 2014, 51, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Vieler, E.; Baumgärtner, W.; Herbst, W.; Köhler, G. Characterization of a reovirus isolate from a rattle snake, Crotalus viridis, with neurological dysfunction. Arch. Virol. 1994, 138, 341–344. [Google Scholar] [CrossRef]
- Yu, K.; Ti, J.; Lu, X.; Pan, L.; Liu, L.; Gao, Y.; Guo, X.; Hu, F.; Liu, C.; Ma, X.; et al. Novel duck reovirus exhibits pathogenicity to specific pathogen-free chickens by the subcutaneous route. Sci. Rep. 2021, 11, 11769. [Google Scholar] [CrossRef]
- Chua, K.B.; Crameri, G.; Hyatt, A.; Yu, M.; Tompang, M.R.; Rosli, J.; McEachern, J.; Crameri, S.; Kumarasamy, V.; Eaton, B.T.; et al. A previously unknown reovirus of bat origin is associated with an acute respiratory disease in humans. Proc. Natl. Acad. Sci. USA 2007, 104, 11424–11429. [Google Scholar] [CrossRef]
- Luo, Y.; Fei, L.; Yue, H.; Li, S.; Ma, H.; Tang, C. Prevalence and genomic characteristics of a novel reassortment mammalian orthoreovirus type 2 in diarrhea piglets in Sichuan, China. Infect. Genet. Evol. 2020, 85, 104420. [Google Scholar] [CrossRef]
- Kwon, H.J.; Kim, H.H.; Kim, H.J.; Park, J.G.; Son, K.Y.; Jung, J.; Lee, W.S.; Cho, K.O.; Park, S.J.; Kang, M.I. Detection and molecular characterization of porcine type 3 orthoreoviruses circulating in South Korea. Vet. Microbiol. 2012, 157, 456–463. [Google Scholar] [CrossRef]
- Wang, L.; Li, Y.; Walsh, T.; Shen, Z.; Li, Y.; Deb Nath, N.; Lee, J.; Zheng, B.; Tao, Y.; Paden, C.R.; et al. Isolation and characterization of novel reassortant mammalian orthoreovirus from pigs in the United States. Emerg. Microbes Infect. 2021, 10, 1137–1147. [Google Scholar] [CrossRef]
- Thimmasandra Narayanappa, A.; Sooryanarain, H.; Deventhiran, J.; Cao, D.; Ammayappan Venkatachalam, B.; Kambiranda, D.; LeRoith, T.; Heffron, C.L.; Lindstrom, N.; Hall, K.; et al. A novel pathogenic Mammalian orthoreovirus from diarrheic pigs and Swine blood meal in the United States. mBio 2015, 6, e00593-15. [Google Scholar] [CrossRef]
- Pawęska, J.T.; Storm, N.; Markotter, W.; Di Paola, N.; Wiley, M.R.; Palacios, G.; Jansen van Vuren, P. Shedding of Marburg Virus in Naturally Infected Egyptian Rousette Bats, South Africa, 2017. Emerg. Infect. Dis. 2020, 26, 3051–3055. [Google Scholar] [CrossRef] [PubMed]
- Field, H.E. Hendra virus ecology and transmission. Curr. Opin. Virol. 2016, 16, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Aditi; Shariff, M. Nipah virus infection: A review. Epidemiol. Infect. 2019, 147, e95. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef]
- He, B.; Huang, X.; Zhang, F.; Tan, W.; Matthijnssens, J.; Qin, S.; Xu, L.; Zhao, Z.; Yang, L.; Wang, Q.; et al. Group A Rotaviruses in Chinese Bats: Genetic Composition, Serology, and Evidence for Bat-to-Human Transmission and Reassortment. J. Virol. 2017, 91, e02493-16. [Google Scholar] [CrossRef]
- He, B.; Yang, F.; Yang, W.; Zhang, Y.; Feng, Y.; Zhou, J.; Xie, J.; Feng, Y.; Bao, X.; Guo, H.; et al. Characterization of a novel G3P[3] rotavirus isolated from a lesser horseshoe bat: A distant relative of feline/canine rotaviruses. J. Virol. 2013, 87, 12357–12366. [Google Scholar] [CrossRef]
- Harima, H.; Sasaki, M.; Orba, Y.; Okuya, K.; Qiu, Y.; Wastika, C.E.; Changula, K.; Kajihara, M.; Simulundu, E.; Yamaguchi, T.; et al. Attenuated infection by a Pteropine orthoreovirus isolated from an Egyptian fruit bat in Zambia. PLoS Negl. Trop. Dis. 2021, 15, e0009768. [Google Scholar] [CrossRef]
- Hu, T.; Qiu, W.; He, B.; Zhang, Y.; Yu, J.; Liang, X.; Zhang, W.; Chen, G.; Zhang, Y.; Wang, Y.; et al. Characterization of a novel orthoreovirus isolated from fruit bat, China. BMC Microbiol. 2014, 14, 293. [Google Scholar] [CrossRef]
- Du, L.; Lu, Z.; Fan, Y.; Meng, K.; Jiang, Y.; Zhu, Y.; Wang, S.; Gu, W.; Zou, X.; Tu, C. Xi River virus, a new bat reovirus isolated in southern China. Arch. Virol. 2010, 155, 1295–1299. [Google Scholar] [CrossRef]
- Pritchard, L.I.; Chua, K.B.; Cummins, D.; Hyatt, A.; Crameri, G.; Eaton, B.T.; Wang, L.F. Pulau virus; a new member of the Nelson Bay orthoreovirus species isolated from fruit bats in Malaysia. Arch. Virol. 2006, 151, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Sheng, J.; Zhao, Z.; Yan, C.; Tu, C.; He, B. Genomic Characterization of the First Parechovirus in Bats. Virol. Sin. 2019, 34, 471–473. [Google Scholar] [CrossRef]
- Tan, Z.; Gonzalez, G.; Sheng, J.; Wu, J.; Zhang, F.; Xu, L.; Zhang, P.; Zhu, A.; Qu, Y.; Tu, C.; et al. Extensive Genetic Diversity of Polyomaviruses in Sympatric Bat Communities: Host Switching versus Coevolution. J. Virol 2020, 94, e02101-19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Yan, C.; Zhao, Z.; Tu, C.; Qu, Y.; Sheng, J.; He, B. Viral metagenomic analyses of bats in Xinjiang, China. Chin. J. Virol. 2018, 34, 896–903. [Google Scholar]
- Zhang, S.; Yang, F.; Yu, L.; He, B.; Tu, C. Immortalization of bat fetal kidney cell by SV40 large T antigen. Chin. J. Prev. Vet. Med. 2014, 36, 363. [Google Scholar]
- Sun, Y.; Qu, Y.; Yan, X.; Yan, G.; Chen, J.; Wang, G.; Zhao, Z.; Liu, Y.; Tu, C.; He, B. Comprehensive Evaluation of RNA and DNA Viromic Methods Based on Species Richness and Abundance Analyses Using Marmot Rectal Samples. mSystems 2022, e0043022. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yan, X.; Sun, Y.; Ren, Z.; Yan, G.; Wang, G.; Liu, Y.; Zhao, Z.; Liu, Y.; Tu, C.; et al. De-heterogeneity of the eukaryotic viral reference database (EVRD) improves the accuracy and efficiency of viromic analysis. bioRxiv 2022. [Google Scholar] [CrossRef]
- Rombel, I.T.; Sykes, K.F.; Rayner, S.; Johnston, S.A. ORF-FINDER: A vector for high-throughput gene identification. Gene 2002, 282, 33–41. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Higgins, D.G. Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2002, 1, 2–3. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Khoosal, A.; Muhire, B. Detecting and Analyzing Genetic Recombination Using RDP4. Methods Mol. Biol. 2017, 1525, 433–460. [Google Scholar] [PubMed]
- Samson, S.; Lord, É.; Makarenkov, V. SimPlot ++: A Python application for representing sequence similarity and detecting recombination. Bioinformatics 2022, 38, 3118–3120. [Google Scholar] [CrossRef]
- Bezzaouha, A.; Bouamra, A.; Ammimer, A.; Ben Abdelaziz, A. Non-parametric tests on SPSS to compare two or more means on matched samples. Tunis Med. 2020, 98, 932–941. [Google Scholar]
- Chandran, K.; Nibert, M.L. Protease cleavage of reovirus capsid protein mu1/mu1C is blocked by alkyl sulfate detergents, yielding a new type of infectious subvirion particle. J. Virol. 1998, 72, 467–475. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, L. Fields Virology, 6th Edition. Clin. Infect. Dis. 2014, 59, 613. [Google Scholar] [CrossRef]
- Mikuletič, T.; Steyer, A.; Kotar, T.; Zorec, T.M.; Poljak, M. A novel reassortant mammalian orthoreovirus with a divergent S1 genome segment identified in a traveler with diarrhea. Infect. Genet. Evol. 2019, 73, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Kohl, C.; Lesnik, R.; Brinkmann, A.; Ebinger, A.; Radonić, A.; Nitsche, A.; Mühldorfer, K.; Wibbelt, G.; Kurth, A. Isolation and characterization of three mammalian orthoreoviruses from European bats. PLoS ONE 2012, 7, e43106. [Google Scholar] [CrossRef]
- Lelli, D.; Moreno, A.; Steyer, A.; Nagliˇc, T.; Chiapponi, C.; Prosperi, A.; Faccin, F.; Sozzi, E.; Lavazza, A. Detection and Characterization of a Novel Reassortant Mammalian Orthoreovirus in Bats in Europe. Viruses 2015, 7, 5844–5854. [Google Scholar] [CrossRef]
- Naglič, T.; Rihtarič, D.; Hostnik, P.; Toplak, N.; Koren, S.; Kuhar, U.; Jamnikar-Ciglenečki, U.; Kutnjak, D.; Steyer, A. Identification of novel reassortant mammalian orthoreoviruses from bats in Slovenia. BMC Vet. Res. 2018, 14, 264. [Google Scholar] [CrossRef]
- Lewandowska, D.W.; Capaul, R.; Prader, S.; Zagordi, O.; Geissberger, F.D.; Kügler, M.; Knorr, M.; Berger, C.; Güngör, T.; Reichenbach, J.; et al. Persistent mammalian orthoreovirus, coxsackievirus and adenovirus co-infection in a child with a primary immunodeficiency detected by metagenomic sequencing: A case report. BMC Infect. Dis. 2018, 18, 33. [Google Scholar] [CrossRef]
- Li, Z.; Shao, Y.; Liu, C.; Liu, D.; Guo, D.; Qiu, Z.; Tian, J.; Zhang, X.; Liu, S.; Qu, L. Isolation and pathogenicity of the mammalian orthoreovirus MPC/04 from masked civet cats. Infect. Genet. Evol. 2015, 36, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.L.; Tan, B.; Wang, B.; Li, W.; Wang, N.; Luo, C.M.; Wang, M.N.; Zhang, W.; Li, B.; Peng, C.; et al. Isolation and identification of bat viruses closely related to human, porcine and mink orthoreoviruses. J. Gen. Virol. 2015, 96, 3525–3531. [Google Scholar] [CrossRef] [PubMed]
- Ahasan, M.S.; Subramaniam, K.; Sayler, K.A.; Loeb, J.C.; Popov, V.L.; Lednicky, J.A.; Wisely, S.M.; Campos Krauer, J.M.; Waltzek, T.B. Molecular characterization of a novel reassortment Mammalian orthoreovirus type 2 isolated from a Florida white-tailed deer fawn. Virus Res. 2019, 270, 197642. [Google Scholar] [CrossRef]
- Richardson, S.C.; Bishop, R.F.; Smith, A.L. Reovirus serotype 3 infection in infants with extrahepatic biliary atresia or neonatal hepatitis. J. Gastroenterol. Hepatol. 1994, 9, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Lelli, D.; Beato, M.S.; Cavicchio, L.; Lavazza, A.; Chiapponi, C.; Leopardi, S.; Baioni, L.; De Benedictis, P.; Moreno, A. First identification of mammalian orthoreovirus type 3 in diarrheic pigs in Europe. Virol. J. 2016, 13, 139. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.D.; Li, B.; Liu, X.L.; Liu, M.Q.; Chen, J.; Luo, D.S.; Hu, B.J.; Zhang, W.; Li, S.Y.; Yang, X.L.; et al. Bat mammalian orthoreoviruses cause severe pneumonia in mice. Virology 2020, 551, 84–92. [Google Scholar] [CrossRef]
- Qin, P.; Li, H.; Wang, J.W.; Wang, B.; Xie, R.H.; Xu, H.; Zhao, L.Y.; Li, L.; Pan, Y.; Song, Y.; et al. Genetic and pathogenic characterization of a novel reassortant mammalian orthoreovirus 3 (MRV3) from a diarrheic piglet and seroepidemiological survey of MRV3 in diarrheic pigs from east China. Vet. Microbiol. 2017, 208, 126–136. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| County | Tissues | Individuals | ||||
|---|---|---|---|---|---|---|
| Brains | Livers | Kidneys | Lungs | Intestines | ||
| Qapqal | 7.9% (6/76) | 2.6% (2/76) | 7.9% (6/76) | 9.2% (7/76) | 14.5% (11/76) | 26.3% (20/76) |
| Xinyuan | 0% (0/46) | 6.5% (3/46) | 0% (0/46) | 2.1% (1/46) | 4.3% (2/46) | 10.9% (5/46) |
| Segments | Similarity | Strains | Host | Disease | Country | Years | Accession No. |
|---|---|---|---|---|---|---|---|
| L1 | 97.0% | YNSZ/V207/2016 | Cattle | NA | China | 2016 | MW198704.1 |
| L2 | 95.1% | SI-MRV07 | Human | Diarrhea | Slovenia | 2017 | MG999577.1 |
| L3 | 93.6–95.9% | SI-MRV07 | Human | Diarrhea | Slovenia | 2017 | MG999578.1 |
| M1 | 93.8% | mew716 | Human | Flu-like, diarrhea | Switzerland | 2015 | KX932032.1 |
| M2 | 98.0–98.1% | WIV4 | Bat. | NA | China | 2011 | KT444536.1 |
| M3 | 98.1–98.2% | SI-MRV04 | Bat | NA | Slovenia | 2009 | MG457103.1 |
| S1 | 92.4% | OV204 | Deer | Lethargy | USA | 2016 | MK092970.1 |
| S2 | 95.7–95.9% | SI-MRV07 | Human | Diarrhea | Slovenia | 2017 | MG999583.1 |
| S3 | 91.2–91.3% | MPC/04 | Civet | Diarrhea | China | 2004 | GQ468274.1 |
| S4 | 97.8–97.9% | WIV5 | Bat | NA | China | 2011 | KT444551.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yan, X.; Sheng, J.; Zhang, C.; Li, N.; Yi, L.; Zhao, Z.; Feng, Y.; Tu, C.; He, B. Detection and Characterization of a Reassortant Mammalian Orthoreovirus Isolated from Bats in Xinjiang, China. Viruses 2022, 14, 1897. https://doi.org/10.3390/v14091897
Yan X, Sheng J, Zhang C, Li N, Yi L, Zhao Z, Feng Y, Tu C, He B. Detection and Characterization of a Reassortant Mammalian Orthoreovirus Isolated from Bats in Xinjiang, China. Viruses. 2022; 14(9):1897. https://doi.org/10.3390/v14091897
Chicago/Turabian StyleYan, Xiaomin, Jinliang Sheng, Chang Zhang, Nan Li, Le Yi, Zihan Zhao, Ye Feng, Changchun Tu, and Biao He. 2022. "Detection and Characterization of a Reassortant Mammalian Orthoreovirus Isolated from Bats in Xinjiang, China" Viruses 14, no. 9: 1897. https://doi.org/10.3390/v14091897
APA StyleYan, X., Sheng, J., Zhang, C., Li, N., Yi, L., Zhao, Z., Feng, Y., Tu, C., & He, B. (2022). Detection and Characterization of a Reassortant Mammalian Orthoreovirus Isolated from Bats in Xinjiang, China. Viruses, 14(9), 1897. https://doi.org/10.3390/v14091897