
Pathogenic Connections in Post-COVID Conditions: What Do We Know in the Large Unknown? A Narrative Review

 ,
,  ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
3. Epidemiology and Pathogenic Factors in MIS
4. Comorbidities and Cofactors
5. Immune and Hematological Implications
6. Cardiovascular Complications
7. Endocrine and Metabolic Considerations in PCC
7.1. COVID-19 and Thyroid Disorders
7.2. COVID-19 and Diabetes
7.3. Hypocalcemia
7.4. Low Levels of Vitamin D
7.5. COVID-19 and Hypothalamus–Pituitary Axis Disorders
7.6. COVID-19 and Gonads
7.7. COVID-19 and the Syndrome of Inappropriate Antidiuretic Hormone Secretion (SIADH)
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sood, M.; Sharma, S.; Sood, I.; Sharma, K.; Kaushik, A. Emerging evidence on multisystem inflammatory syndrome in children associated with SARS-CoV-2 infection: A systematic review with meta-analysis. SN Compr. Clin. Med. 2021, 7, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Nakra, N.A.; Blumberg, D.A.; Herrera-Guerra, A.; Lakshminrusimha, S. Multi-system inflammatory syndrome in children (MIS-C) following SARS-CoV-2 infection: Review of clinical presentation, hypothetical pathogenesis, and proposed management. Children 2020, 7, 69. [Google Scholar] [CrossRef]
- Ramos-Casals, M.; Brito-Zerón, P.; Mariette, X. Systemic and organ-specific immune-related manifestations of COVID-19. Nat. Rev. Rheumatol. 2021, 17, 315–332. [Google Scholar] [CrossRef] [PubMed]
- Esfandyarpour, R.; Kashi, A.; Nemat-Gorgani, M.; Wilhelmy, J.; Davis, R.W. A nanoelectronics-blood-based diagnostic biomarker for myalgic encephalomyelitis/chronic fatigue syndrome (ME/CFS). Proc. Natl. Acad. Sci. USA 2019, 116, 10250–10257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Huang, L.; Wang, Y.; Li, X.; Ren, L.; Gu, X.; Kang, L.; Guo, L.; Liu, M.; Zhou, X.; et al. 6-month consequences of COVID-19 in patients discharged from hospital: A cohort study. Lancet 2021, 397, 220–232. [Google Scholar] [CrossRef]
- Chopra, V.; Flanders, S.A.; O’Malley, M.; Malani, A.N.; Prescott, H.C. Sixty-day outcomes among patients hospitalized with COVID-19. Ann. Intern. Med. 2021, 174, 576–578. [Google Scholar] [CrossRef] [PubMed]
- Carfì, A.; Bernabei, R.; Landi, F. Gemelli against COVID-19 post-acute care study group. Persistent symptoms in patients after acute COVID-19. JAMA 2020, 324, 603–605. [Google Scholar] [CrossRef] [PubMed]
- Crook, H.; Raza, S.; Nowell, J.; Young, M.; Edison, P. Long covid-mechanisms, risk factors, and management. BMJ 2021, 374, n1648. [Google Scholar] [CrossRef]
- Moreno-Pérez, O.; Merino, E.; Leon-Ramirez, J.-M.; Andres, M.; Ramos, J.M.; Arenas-Jiménez, J.; Asensio, S.; Sanchez, R.; Ruiz-Torregrosa, P.; Galan, I.; et al. Post-acute COVID-19 syndrome. Incidence and risk factors: A Mediterranean cohort study. J. Infect. 2021, 82, 378–383. [Google Scholar] [CrossRef]
- Hope, A.A.; Evering, T.H. Post-acute sequelae of SARS CoV-2 infection. Infect. Dis. Clin. N. Am. 2022, 36, 379–395. [Google Scholar] [CrossRef] [PubMed]
- Riphagen, S.; Gomez, X.; Gonzalez-Martinez, C.; Wilkinson, N.; Theocharis, P. Hyperinflammatory shock in children during COVID-19 pandemic. Lancet 2020, 395, 1607–1608. [Google Scholar] [CrossRef]
- Jiang, L.; Tang, K.; Levin, M.; Irfan, O.; Morris, S.K.; Wilson, K.; Klein, J.D.; Bhutta, Z.A. COVID-19 and multisystem inflammatory syndrome in children and adolescents. Lancet Infect. Dis. 2020, 20, e276–e288. [Google Scholar] [CrossRef]
- Henderson, L.; Canna, S.; Friedman, K.; Gorelik, M.; Lapidus, S.K.; Bassiri, H.; Behrens, E.M.; Ferris, A.; Kernan, K.F.; Schulert, G.S.; et al. American College of Rheumatology clinical guidance for multisystem inflammatory syndrome in children associated with SARS-CoV-2 and hyperinflammation in pediatric COVID-19: Version 3. Arthritis Rheumatol. 2022, 74, e1–e20. [Google Scholar] [CrossRef] [PubMed]
- Buonsenso, D.; Munblit, D.; De Rose, C.; Sinatti, D.; Ricchiuto, A.; Carfi, A.; Valentini, P. Preliminary evidence on long COVID in children. Acta Paediatr. 2021, 110, 2208–2211. [Google Scholar] [CrossRef]
- Garg, M.; Maralakunte, M.; Garg, S.; Dhooria, S.; Sehgal, I.; Bhalla, A.S.; Vijayvergiya, R.; Grover, S.; Bhatia, V.; Jagia, P.; et al. The conundrum of ‘Long-COVID-19′: A narrative review. Int. J. Gen. Med. 2021, 14, 2491–2506. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Gómez, H.R.; Morfín-Otero, R.; González-Díaz, E.; Esparza-Ahumada, S.; León-Garnica, G.; Rodríguez-Noriega, E. The multifaceted manifestations of multisystem inflammatory syndrome during the SARS-CoV-2 pandemic. Pathogens 2022, 11, 556. [Google Scholar] [CrossRef]
- Trapani, G.; Verlato, G.; Bertino, E.; Maiocco, G.; Vesentini, R.; Spadavecchia, A.; Dessì, A.; Fanos, V. Long COVID-19 in children: An Italian cohort study. Ital. J. Pediatr. 2022, 48, 83. [Google Scholar] [CrossRef] [PubMed]
- Asadi-Pooya, A.A.; Nemati, H.; Shahisavandi, M.; Akbari, A.; Emami, A.; Lotfi, M.; Rostamihosseinkhani, M.; Barzegar, Z.; Kabiri, M.; Zeraatpisheh, Z.; et al. Long COVID in children and adolescents. World J. Pediatr. 2021, 17, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Williams, V.; Dash, N.; Suthar, R.; Mohandoss, V.; Jaiswal, N.; Kavitha, T.K.; Nallasamy, K.; Angurana, S.K. Clinicolaboratory profile, treatment, intensive care needs, and outcome of pediatric inflammatory multisystem syndrome temporally associated with SARS-CoV-2: A systematic review and meta-analysis. J. Pediatr. Intensive Care 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Rowley, A.H. Understanding SARS-CoV-2-related multisystem inflammatory syndrome in children. Nat. Rev. Immunol. 2020, 20, 453–454. [Google Scholar] [CrossRef]
- Parums, D.W. Multisystem inflammatory syndrome in adults (MIS-A) and the spectrum of COVID-19. Med. Sci. Monit. 2021, 27, e935005. [Google Scholar] [CrossRef] [PubMed]
- Kunal, S.; Ish, P.; Sakthivel, P.; Malhotra, N.; Gupta, K. The emerging threat of multisystem inflammatory syndrome in adults (MIS-A) in COVID-19: A systematic review. Heart Lung 2022, 54, 7–18. [Google Scholar] [CrossRef]
- Feldstein, L.R.; Tenforde, M.W.; Friedman, K.G.; Newhams, M.; Rose, E.B.; Dapul, H.; Soma, V.L.; Maddux, A.B.; Mourani, P.M.; Bowens, C.; et al. Characteristics and outcomes of US children and adolescents with multisystem inflammatory syndrome in children (MIS-C) compared with severe acute COVID-19. JAMA 2021, 325, 1074–1087. [Google Scholar] [CrossRef] [PubMed]
- Sil, A.; Das, A.; Datta, D. Mucocutaneous manifestations of COVID-19 related multisystem inflammatory syndrome in adults (MIS-A): An update. Clin. Exp. Dermatol. 2022. [Google Scholar] [CrossRef] [PubMed]
- Maltezou, H.C.; Pavli, A.; Tsakris, A. Post-COVID syndrome: An insight on its pathogenesis. Vaccines 2021, 9, 497. [Google Scholar] [CrossRef]
- Zeichner, S.L.; Cruz, A.T. Multisystem inflammatory syndrome in children and SARS-CoV-2 serology. Pediatrics 2020, 146, e2020032888. [Google Scholar] [CrossRef] [PubMed]
- Merckx, J.; Cooke, S.; El Tal, T.; Bitnun, A.; Morris, S.K.; Yeh, E.A.; Yea, C.; Gill, P.; Papenburg, J.; Lefebvre, M.A.; et al. Predictors of severe illness in children with multisystem inflammatory syndrome after SARS-CoV-2 infection: A multicentre cohort study. CMAJ 2022, 194, E513–E523. [Google Scholar] [CrossRef] [PubMed]
- Cirks, B.T.; Rowe, S.J.; Jiang, S.Y.; Brooks, R.M.; Mulreany, M.P.; Hoffner, W.; Jones, O.Y.; Hickey, P.W. Sixteen weeks later: Expanding the risk period for multisystem inflammatory syndrome in children. J. Pediatr. Infect. Dis. Soc. 2021, 10, 686–690. [Google Scholar] [CrossRef]
- Vogel, T.P.; Top, K.A.; Karatzios, C.; Hilmers, D.C.; Tapia, L.I.; Moceri, P.; Giovannini-Chami, L.; Wood, N.; Chandler, R.E.; Klein, N.P.; et al. Multisystem inflammatory syndrome in children and adults (MIS-C/A): Case definition and guidelines for data collection, analysis, and presentation of immunization safety data. Vaccine 2021, 39, 3037–3049. [Google Scholar] [CrossRef] [PubMed]
- Abrams, J.Y.; Oster, M.; Godfred-Cato, S.; Bryant, B.; Datta, S.D.; Campbell, A.P.; Leung, J.W.; Tsang, C.; Pierce, T.J.; Kennedy, J.L.; et al. Factors linked to severe outcomes in multisystem inflammatory syndrome in children (MIS-C) in the USA: A retrospective surveillance study. Lancet Child Adolesc. Health 2021, 5, 323–331. [Google Scholar] [CrossRef]
- Feldstein, L.R.; Rose, E.B.; Horwitz, S.M.; Collins, J.P.; Newhams, M.M.; Son, M.B.F.; Newburger, J.W.; Kleinman, L.C.; Heidemann, S.M.; Martin, A.A.; et al. Multisystem inflammatory syndrome in U.S. children and adolescents. N. Engl. J. Med. 2020, 383, 334–346. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.Y.; Xu, B.W.; Du, J.B. Similarities and differences between multiple inflammatory syndrome in children associated with COVID-19 and Kawasaki disease: Clinical presentations, diagnosis, and treatment. World J. Pediatr. 2021, 17, 335–340. [Google Scholar] [CrossRef]
- Fernández-Sarmiento, J.; De Souza, D.; Jabornisky, R.; Gonzalez, G.A.; López, M.D.P.A.; Palacio, G. Pediatric inflammatory multisystem syndrome temporally associated with COVID-19 (PIMS-TS): A narrative review and the viewpoint of the Latin American Society of Pediatric Intensive Care (SLACIP) Sepsis Committee. BMJ Paediatr. Open 2021, 5, e000894. [Google Scholar] [CrossRef] [PubMed]
- Boeckelmann, M.; Glaser, N.; Dejas, F.; Östreicher, I.; Grüner, J.; Höche, A.; Akanbi, S.; Thiemig, D.; Rossi, R. Pediatric inflammatory multisystem syndrome-Erfahrungen aus einer Berliner Kinderklinik. Mon. Kinderheilkd 2022, 170, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Ng, K.F.; Kothari, T.; Bandi, S.; Bird, P.W.; Goyal, K.; Zoha, M.; Rai, V.; Tang, J.W. COVID-19 multisystem inflammatory syndrome in three adolescents with confirmed SARS-CoV-2 infection. J. Med. Virol. 2020, 92, 2880–2886. [Google Scholar] [CrossRef] [PubMed]
- Bautista-Rodriguez, C.; Sanchez-de-Toledo, J.; Clark, B.C.; Herberg, J.; Bajolle, F.; Randanne, P.C.; Salas-Mera, D.; Foldvari, S.; Chowdhury, D.; Munoz, R.; et al. Multisystem inflammatory syndrome in children: An international survey. Pediatrics 2021, 147, 2. [Google Scholar] [CrossRef]
- Wang, Y.; Deng, H.; Pan, L.; Hu, R.; Lu, Y.; Deng, W.; Sun, W.; Chen, C.; Shen, X.; Huang, X.F. Periodontal diseases increases the host susceptibility to COVID-19 and its severity: A Mendelian randomization study. J. Transl. Med. 2021, 19, 528. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Mohindra, R.; Singla, M.; Khera, S.; Sahni, V.; Kanta, P.; Soni, R.K.; Kumar, A.; Gauba, K.; Goyal, K.; et al. The clinical association between periodontitis and COVID-19. Clin. Oral Investig. 2021, 27, 1361–1374. [Google Scholar] [CrossRef] [PubMed]
- Marouf, N.; Cai, W.; Said, K.N.; Daas, H.; Diab, H.; Chinta, V.R.; Hssain, A.A.; Nicolau, B.; Sanz, M.; Tamimi, F. Association between periodontitis and severity of COVID-19 infection: A case-control study. J. Clin. Periodontol. 2021, 48, 483–491. [Google Scholar] [CrossRef]
- Basso, L.; Chacun, D.; Sy, K. Periodontal diseases and COVID-19: A scoping review. Eur. J. Dent. 2021, 15, 768–775. [Google Scholar] [CrossRef]
- Botros, N.; Iyer, P.; Ojcius, D.M. Is there an association between oral health and severity of COVID-19 complications? Biomed. J. 2020, 43, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Bourgeois, D.; Inquimbert, C.; Ottolenghi, L.; Carrouel, F. Periodontal pathogens as risk factors of cardiovascular diseases, diabetes, rheumatoid arthritis, cancer, and chronic obstructive pulmonary disease—Is there cause for consideration? Microorganisms 2019, 7, 424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raisanen, I.T.; Umeizudike, K.A.; Partanen, P.; Heikkilä, P.; Tervahartiala, T.; Nwhator, S.O.; Grigoriadis, A.; Sakellari, D.; Sorsa, T. Periodontal disease and targeted prevention using aMMP-8 point-of-care oral fluid analytics in the COVID-19 era. Med. Hypotheses 2020, 144, 110276. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Watanabe, N.; Kamio, N.; Kobayashi, R.; Iinuma, T.; Imai, K. Aspiration of periodontopathic bacteria due to poor oral hygiene potentially contributes to the aggravation of COVID-19. J. Oral Sci. 2020, 63, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.; Xiao, Y.; Kang, L.; Ma, W.; Shi, L.; Zhang, L.; Zhou, Z.; Yang, J.; Zhong, J.; Yang, D.; et al. Genomic diversity of severe acute respiratory syndrome-coronavirus 2 in patients with coronavirus disease 2019. Clin. Infect. Dis. 2020, 71, 713–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, J.G.; Yoon, J.; Song, J.Y.; Yoon, S.Y.; Lim, C.S.; Seong, H.; Noh, J.Y.; Cheong, H.J.; Kim, W.J. Clinical significance of a high SARS-CoV-2 viral load in the saliva. J. Korean Med. Sci. 2020, 35, e195. [Google Scholar] [CrossRef]
- Espinoza-Espinoza, D.A.; Dulanto-Vargas, J.A.; Cáceres-LaTorre, O.A.; Lamas-Castillo, F.E.; Flores-Mir, C.; Cervantes-Ganoza, L.A.; López-Gurreonero, C.; Ladera-Castañeda, M.I.; Cayo-Rojas, C.F. Association between periodontal disease and the risk of COVID-19 complications and mortality: A systematic review. J. Int. Soc. Prevent. Communit. Dent. 2021, 11, 626–638. [Google Scholar]
- Preshaw, P.; Bissett, S. Periodontitis and diabetes. Br. Dent. J. 2019, 227, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Herrera, M.; Silvestre-Rangil, J.; Silvestre, F.J. Association between obesity and periodontal disease. A systematic review of epidemiological studies and controlled clinical trials. Med. Oral Patol. Oral Cir. Bucal. 2017, 22, e708–e715. [Google Scholar] [CrossRef]
- Hasan, L.K.; Deadwiler, B.; Haratian, A.; Bolia, I.K.; Weber, A.E.; Petrigliano, F.A. Effects of COVID-19 on the musculoskeletal system: Clinician’s guide. Orthop. Res. Rev. 2021, 13, 141–150. [Google Scholar] [CrossRef]
- Sanz, M.; Herrera, D.; Kebschull, M.; Chapple, I.; Jepsen, S.; Beglundh, T.; Sculean, A.; Tonetti, M.S.; EFP Workshop Participants and Methodological Consultants. Treatment of stage I-III periodontitis-The EFP S3 level clinical practice guideline. J. Clin. Periodontol. 2020, 47, 4–60. [Google Scholar] [CrossRef] [PubMed]
- Abou-Bakr, A.; Hussein, R.R.; Khalil, E.; Ahmed, E. The frequency of periodontitis in end-stage renal disease on hemodialysis in a sample of Egyptian population: Multi-center clinical cross-sectional study. BMC Oral Health 2022, 22, 1. [Google Scholar] [CrossRef] [PubMed]
- Sanz, M.; Marco Del Castillo, A.; Jepsen, S.; Gonzalez-Juanatey, J.R.; D’Aiuto, F.; Bouchard, P.; Chapple, I.; Dietrich, T.; Gotsman, I.; Graziani, F.; et al. Periodontitis and cardiovascular diseases: Consensus report. J. Clin. Periodontol. 2020, 47, 268–288. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Liu, Y.; Li, Y.; Xu, D.; Zhang, J.; Peng, Z. Severe COVID-19: Immunosuppression or Hyperinflammation? Shock 2021, 56, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Fletman, E.W.; Stumpf, N.; Kalimullah, J.; Levinson, N.; Deboo, A. Guillain-Barré syndrome associated with COVID-19: An atypical, late-onset presentation. Neurol. Sci 2021, 42, 4393–4395. [Google Scholar] [CrossRef] [PubMed]
- Dhanalakshmi, K.; Venkataraman, A.; Balasubramanian, S.; Madhusudan, M.; Amperayani, S.; Putilibai, S.; Sadasivam, K.; Ramachandran, B.; Ramanan, A.V. Epidemiological and clinical profile of pediatric inflammatory multisystem syndrome-temporally associated with SARS-CoV-2 (PIMS-TS) in indian children. Indian Pediatr. 2020, 57, 1010–1014. [Google Scholar] [CrossRef] [PubMed]
- Sedaghat, Z.; Karimi, N. Guillain Barre syndrome associated with COVID-19 infection: A case report. J. Clin. Neurosci. 2020, 76, 233–235. [Google Scholar] [CrossRef] [PubMed]
- Terpos, E.; Ntanasis-Stathopoulos, I.; Elalamy, I.; Kastritis, E.; Sergentanis, T.N.; Politou, M.; Psaltopoulou, T.; Gerotziafas, G.; Dimopoulos, M.A. Hematological findings and complications of COVID-19. Am. J. Hematol. 2020, 95, 834–847. [Google Scholar] [CrossRef] [Green Version]
- Pelle, M.C.; Tassone, B.; Ricchio, M.; Mazzitelli, M.; Davoli, C.; Procopio, G.; Cancelliere, A.; la Gamba, V.; Lio, E.; Matera, G.; et al. Late-onset myocardial infarction and autoimmune haemolytic anemia in a COVID-19 patient without respiratory symptoms, concomitant with a paradoxical increase in inflammatory markers: A case report. J. Med. Case Rep. 2020, 14, 246. [Google Scholar] [CrossRef]
- Bhattacharjee, S.; Banerjee, M. Immune thrombocytopenia secondary to COVID-19: A systematic review. SN Compr. Clin. Med. 2020, 14, 2048–2049. [Google Scholar] [CrossRef] [PubMed]
- Serrano, C.; Espaol, I.; Cascales, A.; Mishra, J.; Barman, B.; Barman, H. Secondary hemophagocytic lymphohistiocytosis in post- COVID-19 patients: A report of two cases. SN Compr. Clin. Med. 2021, 13, 2389–2392. [Google Scholar] [CrossRef] [PubMed]
- Buonsenso, D.; Mariani, F.; Pierri, L.; Morello, R.; Yock-Corrales, A.; Del Aguila, O.; Lazzareschi, I.; Zampino, G.; Nunziata, F.; Valentini, P.; et al. Association between coagulation profile and clinical outcome in children with SARS-CoV-2 infection or MIS-C: A multicenter cross-sectional study. Children 2022, 9, 279. [Google Scholar] [CrossRef] [PubMed]
- Andrade, B.S.; Siqueira, S.; Soares, W.D.A.; Rangel, F.D.S.; Santos, N.; Freitas, A.D.S.; da Silveira, P.R.; Tiwari, S.; Alzahrani, K.; Góes-Neto, A.; et al. Long-COVID and post-COVID health complications: An up-to-date review on clinical conditions and their possible molecular mechanisms. Viruses 2021, 13, 700. [Google Scholar] [CrossRef] [PubMed]
- Akbarialiabad, H.; Taghrir, M.H.; Abdollahi, A.; Ghahramani, N.; Kumar, M.; Paydar, S.; Razani, B.; Mwangi, J.; Asadi-Pooya, A.A.; Malekmakan, L.; et al. Long COVID, a comprehensive systematic scoping review. Infection 2021, 49, 1163–1186. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Liang, X.; Wei, Y.D. Changes in blood coagulation in patients with severe coronavirus disease 2019 (COVID-19): A meta-analysis. Br. J. Haematol. 2020, 189, 1050–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajpal, S.; Tong, M.S.; Borchers, J.; Zareba, K.M.; Obarski, T.P.; Simonetti, O.P.; Daniels, C.J. Cardiovascular magnetic resonance findings in competitive athletes recovering from COVID-19 infection. JAMA Cardiol. 2021, 6, 116–118. [Google Scholar] [CrossRef]
- Aiyegbusi, O.L.; Hughes, S.E.; Turner, G.; Rivera, S.C.; McMullan, C.; Chandan, J.S.; Haroon, S.; Price, G.; Davies, E.H.; Nirantharakumar, K.; et al. Symptoms, complications and management of long COVID: A review. J. R. Soc. Med. 2021, 114, 428–442. [Google Scholar] [CrossRef] [PubMed]
- Eiros, R.; Barreiro-Perez, M.; Martin-Garcia, A.; Almeida, J.; Villacorta, E.; Perez-Pons, A.; Merchan, S.; Torres-Valle, A.; Pablo, C.S.; Gonzalez-Calle, D.; et al. Pericarditis and myocarditis long after SARS-CoV-2 infection: A cross-sectional descriptive study in healthcare workers. MedRxiv 2020. [Google Scholar] [CrossRef]
- Patel, P.; DeCuir, J.; Abrams, J.; Campbell, A.P.; Godfred-Cato, S.; Belay, E.D. Clinical characteristics of multisystem inflammatory syndrome in adults. JAMA Netw. Open 2021, 4, e2126456. [Google Scholar] [CrossRef]
- Pereira, M.F.B.; Litvinov, N.; Farhat, S.C.L.; Eisencraft, A.P.; Gibelli, M.A.B.C.; de Carvalho, W.B.; Fernandes, V.R.; Fink, T.D.T.; Framil, J.V.D.S.; Galleti, K.V.; et al. Severe clinical spectrum with high mortality in pediatric patients with COVID-19 and multisystem inflammatory syndrome. Clinics 2020, 75, e2209. [Google Scholar] [CrossRef]
- Puig-Domingo, M.; Marazuela, M.; Yildiz, B.O.; Giustina, A. COVID-19 and endocrine and metabolic diseases. An updated statement from the European Society of Endocrinology. Endocrine 2021, 72, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.B.; Farzan, M.; Chen, B.; Choe, H. Mechanisms of SARS-CoV-2 entry into cells. Nat. Rev. Mol. Cell Biol. 2022, 23, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Lisco, G.; De Tullio, A.; Stragapede, A.; Solimando, A.; Albanese, F.; Capobianco, M.; Giagulli, V.; Guastamacchia, E.; De Pergola, G.; Vacca, A.; et al. COVID-19 and the Endocrine System: A Comprehensive Review on the Theme. J. Clin. Med. 2021, 10, 2920. [Google Scholar] [CrossRef] [PubMed]
- Bansal, R.; Gubbi, S.; Koch, C.A. COVID-19 and chronic fatigue syndrome: An endocrine perspective. J. Clin. Transl. Endocrinol. 2022, 27, 100284. [Google Scholar] [CrossRef]
- Clarke, S.A.; Abbara, A.; Dhillo, W.S. Impact of COVID-19 on the endocrine system: A mini-review. Endocrinology 2022, 163, bqab203. [Google Scholar] [CrossRef]
- Rotondi, M.; Coperchini, F.; Ricci, G.; Denegri, M.; Croce, L.; Ngnitejeu, S.T.; Villani, L.; Magri, F.; Latrofa, F.; Chiovato, L. Detection of SARS-COV-2 receptor ACE-2 mRNA in thyroid cells: A clue for COVID-19-related subacute thyroiditis. J. Endocrinol. Investig. 2021, 44, 1085–1090. [Google Scholar] [CrossRef]
- Li, M.Y.; Li, L.; Zhang, Y.; Wang, X.S. Expression of the SARS-CoV-2 cell receptor gene ACE2 in a wide variety of human tissues. Infect. Dis. Poverty 2020, 9, 45. [Google Scholar] [CrossRef]
- Lania, A.; Sandri, M.; Cellini, M.; Mirani, M.; Lavezzi, E.; Mazziotti, G. Thyrotoxicosis in patients with COVID-19: The THYRCOV study. Eur. J. Endocrinol. 2020, 183, 381–387. [Google Scholar] [CrossRef]
- Montebello, A. Recurrent Graves’ disease post SARS-CoV-2 infection. BMJ Case Rep. 2021, 4, e244714. [Google Scholar] [CrossRef]
- Caron, P. Thyroid disorders and SARS-CoV-2 infection: From pathophysiological mechanism to patient management. Ann. Endocrinol. 2020, 81, 507–510. [Google Scholar] [CrossRef] [PubMed]
- Gardner, D.; Shoback, D. Greenspan’s Basic & Clinical Endocrinology, 10th ed.; McGraw-Hill Education: New York, NY, USA, 2018. [Google Scholar]
- Klein, I.; Ojamaa, K. Thyroid hormone and the cardiovascular system. N. Engl. J. Med. 2001, 344, 501–509. [Google Scholar] [CrossRef]
- Schmidt, B.M.; Martin, N.; Georgens, A.C.; Tillmann, H.C.; Feuring, M.; Christ, M.; Wehling, M. Nongenomic cardiovascular effects of triiodothyronine in euthyroid male volunteers. J. Clin. Endocrinol. Metabol. 2002, 87, 1681–1686. [Google Scholar] [CrossRef]
- Park, K.; Dai, H.; Ojamaa, K.; Lowenstein, E.; Klein, I.; Sellke, F. The direct vasomotor effect of thyroid hormones on rat skeletal muscle resistance arteries. Anesth. Analg. 1997, 85, 734–738. [Google Scholar] [CrossRef]
- Delitala, A. Subclinical hyperthyroidism and the cardiovascular disease. Horm. Metab. Res. 2017, 49, 723–731. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakazawa, H.; Sakurai, K.; Hamada, N.; Momotani, N.; Ito, K. Management of atrial fibrillation in the post-thyrotoxic state. Am. J. Med. 1982, 72, 903–906. [Google Scholar] [CrossRef]
- Scappaticcio, L.; Pitoia, F.; Esposito, K.; Piccardo, A.; Trimboli, P. Impact of COVID-19 on the thyroid gland: An update. Rev. Endocr. Metab. Disord. 2020, 22, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Duntas, L.; Jonklaas, J. COVID-19 and thyroid Diseases: A bidirectional impact. J. Endocr. Soc. 2021, 5, bvab076. [Google Scholar] [CrossRef]
- Naguib, R. Potential relationships between COVID-19 and the thyroid gland: An update. J. Int. Med. Res. 2022, 50, 3000605221082898. [Google Scholar] [CrossRef]
- Osuna, P.; Udovcic, M.; Sharma, M. Hypothyroidism and the heart. Methodist DeBakey Cardiovasc. J. 2017, 13, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, S.E.; Castellanos-Diaz, J.; Srihari, A.; Kadiyala, S.; Leey-Casella, J.; Ghayee, H.K.; Ogunsakin, A. Subacute thyroiditis and heart failure in a patient presenting with COVID-19. J. Investig. Med. High Impact Case Rep. 2021, 9, 232470962110094. [Google Scholar] [CrossRef] [PubMed]
- Lui, D.; Lee, C.; Chow, W.; Lee, A.C.H.; Tam, A.R.T.; Pang, P.; Ho, T.Y.; Fong, C.H.Y.; Law, C.Y.; Leung, E.K.H.; et al. Long COVID in patients with mild to moderate disease: Do thyroid function and autoimmunity play a role? Endocr. Pract. 2021, 27, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Geslot, A.; Chanson, P.; Caron, P. COVID-19, the thyroid and the pituitary-The real state of play. Ann. Endocrinol. 2022, 83, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Hulme, K.D.; Yan, L.; Marshall, R.J.; Bloxham, C.J.; Upton, K.R.; Hasnain, S.Z.; Bielefeldt-Ohmann, H.; Loh, Z.; Ronacher, K.; Chew, K.Y.; et al. High glucose levels increase influenza-associated damage to the pulmonary epithelial-endothelial barrier. eLife 2020, 9, e56907. [Google Scholar] [CrossRef] [PubMed]
- Di Filippo, L.; Formenti, A.M.; Rovere-Querini, P.; Carlucci, M.; Conte, C.; Ciceri, F.; Zangrillo, A.; Giustina, A. Hypocalcemia is highly prevalent and predicts hospitalization in patients with COVID-19. Endocrine 2020, 68, 475–478. [Google Scholar] [CrossRef]
- Alemzadeh, E.; Alemzadeh, E.; Ziaee, M.; Abedi, A.; Salehiniya, H. The effect of low serum calcium level on the severity and mortality of Covid patients: A systematic review and meta-analysis. Immun. Inflamm. Dis. 2021, 9, 1219–1228. [Google Scholar] [CrossRef]
- Alguwaihes, A.M.; Sabico, S.; Hasanato, R.; Al-Sofiani, M.E.; Megdad, M.; Albader, S.S.; Alsari, M.H.; Alelayan, A.; Alyusuf, E.Y.; Alzahrani, S.H.; et al. Severe vitamin D deficiency is not related to SARS-CoV-2 infection but may increase mortality risk in hospitalized adults: A retrospective case-control study in an arab gulf country. Aging Clin. Exp. Res. 2021, 33, 1415–1422. [Google Scholar] [CrossRef]
- Barrea, L.; Verde, L.; Grant, W.B.; Frias-Toral, E.; Sarno, G.; Vetrani, C.; Ceriani, F.; Garcia-Velasquez, E.; Contreras-Briceño, J.; Savastano, S.; et al. Vitamin D: A Role Also in Long COVID-19? Nutrients 2022, 14, 1625. [Google Scholar] [CrossRef] [PubMed]
- Popov, D.; Hadzhiyanev, A. Pituitary apoplexy associated with COVID-19 infection: Review and a case report. Biotechnol. Biotechnol. Equip. 2022, 36, 75–81. [Google Scholar] [CrossRef]
- Nonglait, P.L.; Naik, R.; Raizada, N. Hypophysitis after COVID-19 Infection. Indian J. Endocrinol. Metab. 2021, 25, 255–256. [Google Scholar]
- Frara, S.; Allora, A.; Castellino, L.; di Filippo, L.; Loli, P.; Giustina, A. COVID-19 and the pituitary. Pituitary 2021, 24, 465–481. [Google Scholar] [CrossRef] [PubMed]
- Gazzaruso, C.; Gola, M.; Karamouzis, I.; Giubbini, R.; Giustina, A. Cardiovascular risk in adult patients with growth hormone (GH) deficiency and following substitution with GH–An update. J. Clin. Endocrinol. Metab. 2014, 99, 18–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popovic, V.; Korbonits, M. Metabolic Syndrome Consequent to Endocrine Disorders; Karger Medical and Scientific Publishers: Basel, Switzerland, 2018; Volume 49, pp. 1–19. [Google Scholar]
- Kuperberg, S.J.; Navetta-Modrov, B. The role of obesity in the immunopathogenesis of COVID-19 respiratory disease and critical illness. Am. J. Respir. Cell Mol. Biol. 2021, 65, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Yuen, K.C.J. Growth hormone deficiency, acromegaly and COVID-19: Transitioning from media reports to knowledge and a growth hormone hypothesis. Growth Horm. IGF Res. 2021, 56, 101363. [Google Scholar] [CrossRef] [PubMed]
- Arcopinto, M.; Salzano, A.; Giallauria, F.; Bossone, E.; Isgaard, J.; Marra, A.M.; Bobbio, E.; Vriz, O.; Åberg, D.N.; Masarone, D.; et al. Growth hormone deficiency is associated with worse cardiac function, physical performance, and outcome in chronic heart failure: Insights from the T.O.S.CA. GHD Study. PLoS ONE 2017, 12, e0170058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elkarow, M.H.; Hamdy, A. A suggested role of human growth hormone in control of the COVID-19 Pandemic. Front. Endocrinol. 2020, 11, 569633. [Google Scholar] [CrossRef] [PubMed]
- Villa, A.; Rizzi, N.; Vegeto, E.; Ciana, P.; Maggi, A. Estrogen accelerates the resolution of inflammation in macrophagic cells. Sci. Rep. 2015, 5, 15224. [Google Scholar] [CrossRef]
- Au, A.; Feher, A.; McPhee, L.; Jessa, A.; Oh, S.; Einstein, G. Estrogens, inflammation and cognition. Front. Neuroendocr. 2016, 40, 87–100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowlton, A.A.; Lee, A.R. Estrogen and the cardiovascular system. Pharmacol. Ther. 2012, 135, 57–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camici, M.; Zuppi, P.; Lorenzini, P.; Scarnnecchia, L.; Pinnetti, C.; Cicalini, S.; Nicastri, E.; Petrpsillo, N.; Palmieri, F.; D’Offizi, G.; et al. Role of testosterone in SARS-CoV-2 infection: A key pathogenic factor and a biomarker for severe pneumonia. Int. J. Infect. Dis. 2021, 108, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, A.M.; Bremner, W.J. Testicular Disorders. In Williams Textbook of Endocrinology, 13th ed.; Melmed, S., Polonsky, K.S., Larsen, P.R., Kronenberg, H.M., Eds.; Elsevier Health Sciences: New York, NY, USA, 2015; pp. 688–777. [Google Scholar]
- Deenadayalu, V.P.; White, R.E.; Stallone, J.N.; Gao, X.; Garcia, A.J. Testosterone relaxes coronary arteries by opening the large-conductance, calcium-activated potassium channel. Am. J. Physiol. Circ. Physiol. 2001, 281, H1720-7. [Google Scholar] [CrossRef] [PubMed]
- Ruige, J.B.; Mahmoud, A.M.; De Bacquer, D.; Kaufman, J.M. Endogenous testosterone and cardiovascular disease in healthy men: A meta-analysis. Heart 2011, 97, 870–875. [Google Scholar] [CrossRef] [PubMed]
- Kearney, M.T.; Fox, K.; Lee, A.J.; Prescott, R.J.; Shah, A.; Batin, P.D.; Baig, W.; Lindsay, S.; Callahan, T.S.; Shell, W.; et al. Predicting death due to progressive heart failure in patients with mild-to-moderate chronic heart failure. J. Am. Coll. Cardiol. 2002, 40, 1801–1808. [Google Scholar] [CrossRef] [Green Version]
- Gheorghe, G.; Ilie, M.; Bungau, S.; Stoian, A.M.P.; Bacalbasa, N.; Diaconu, C.C. Is there a relationship between COVID-19 and hyponatremia? Medicina 2021, 57, 55. [Google Scholar] [CrossRef]
- Papageorgiou, A.N.; Moffatt, M. Bilateral pneumonia and inappropriate secretion of antidiuretic hormone in a premature infant. Can. Med. Assoc. J. 1976, 114, 1119–1120. [Google Scholar]
- Park, S.J.; Shin, J.I. Inflammation and hyponatremia: An underrecognized condition? Korean J. Pediatr. 2013, 56, 519–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yousaf, Z.; Al-Shokri, S.D.; Al-Soub, H.; Mohamed, M.F.H. COVID-19-associated SIADH: A clue in the times of pandemic! Am. J. Physiol. Endocrinol. Metab. 2020, 318, E882–E885. [Google Scholar] [CrossRef]
- Karabag, T.; Kalayci, B.; Sayin, M.R.; Erten, T. Atrioventricular conduction defect associated with severe hyponatremia. Clujul. Medical. 2018, 91, 342–345. [Google Scholar] [CrossRef] [Green Version]
- Amir, M.; Renata, A.; Ratana, L.T. Symptomatic sinus bradycardia due to electrolyte imbalances in syndrome of inappropriate antidiuretic hormone (SIADH) related COVID-19: A case report. BMC Infect. Dis. 2021, 21, 465. [Google Scholar] [CrossRef] [PubMed]
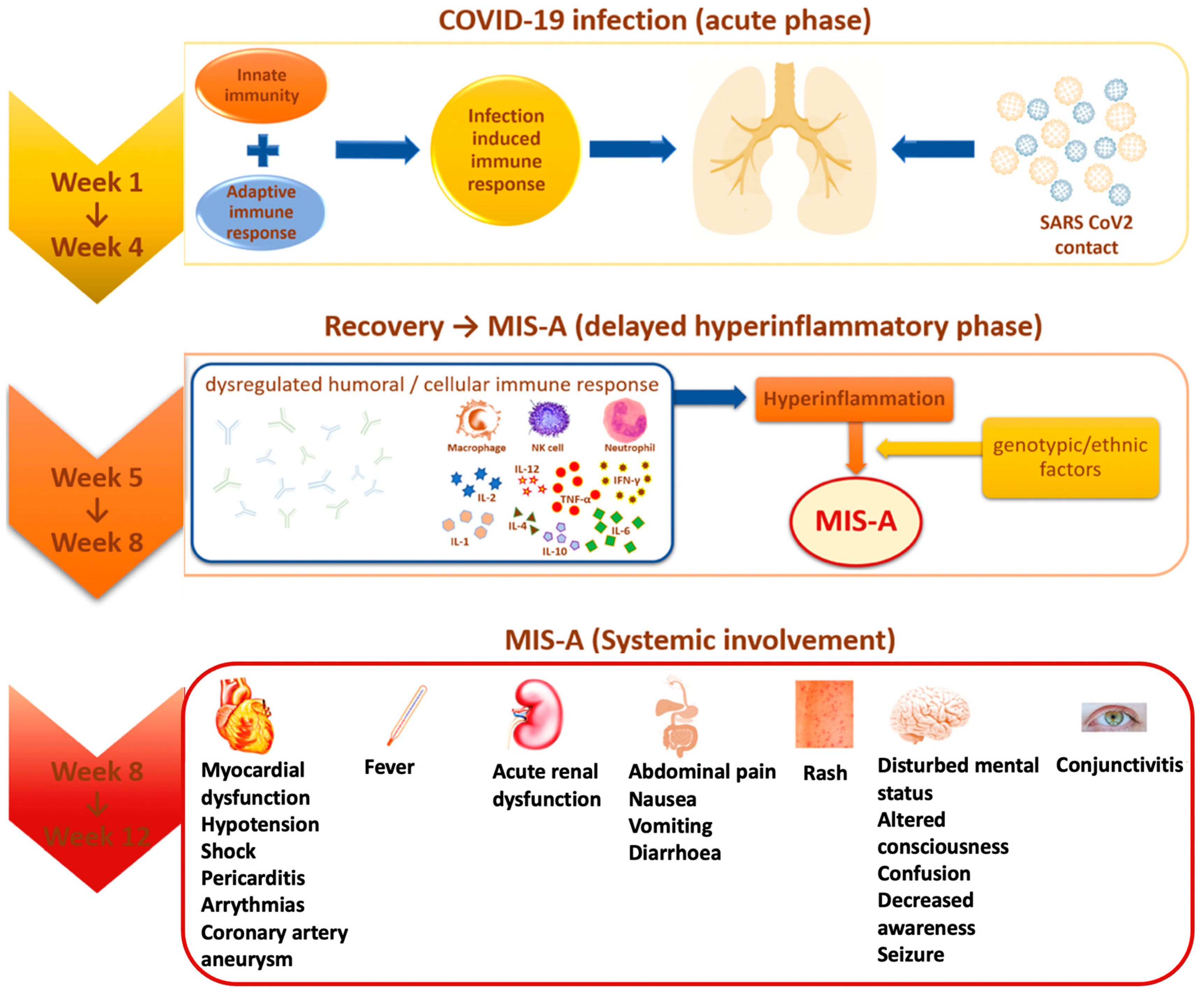

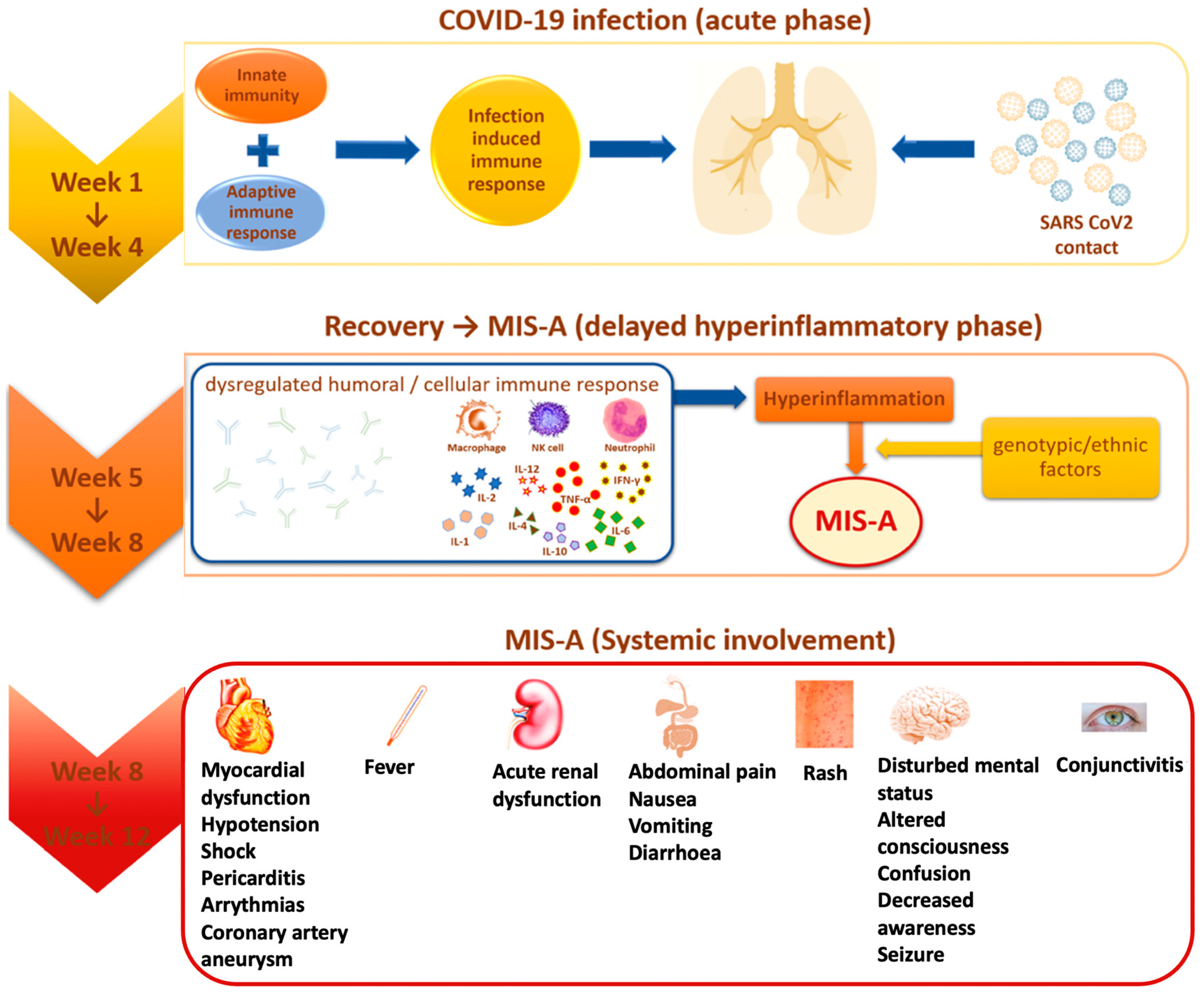{kind=link}
| Diagnostic Criteria | Royal College Criteria | CDC Criteria | WHO Criteria |
|---|---|---|---|
| Age | Not stated | ˂21 years old | 0–19 years old |
| Fever | Persistent fever (≥38.5 °C) | Documented fever ≥ 38 °C or history of continuous fever ≥ 24 h | Fever ≥ 3 days |
| Clinical changes | Both criteria required:
| Both criteria required:
| At least two criteria:
|
| Inflammation markers | All of the following criteria must be present:
| Modification of one or more laboratory tests meaning inflammation (but not limited to)
| The presence of markers of inflammation, for example:
|
| Epidemiological link with SARS-CoV-2 infection | Positive or negative RT-PCR | Current or recent infection sustained by any of the following:
| SARS-CoV-2 infection sustained by any of the following:
|
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stafie, C.S.; Solomon, S.M.; Sufaru, I.-G.; Manaila, M.; Stafie, I.I.; Melinte, G.; Simionescu, B.; Leustean, L. Pathogenic Connections in Post-COVID Conditions: What Do We Know in the Large Unknown? A Narrative Review. Viruses 2022, 14, 1686. https://doi.org/10.3390/v14081686
Stafie CS, Solomon SM, Sufaru I-G, Manaila M, Stafie II, Melinte G, Simionescu B, Leustean L. Pathogenic Connections in Post-COVID Conditions: What Do We Know in the Large Unknown? A Narrative Review. Viruses. 2022; 14(8):1686. https://doi.org/10.3390/v14081686
Chicago/Turabian StyleStafie, Celina Silvia, Sorina Mihaela Solomon, Irina-Georgeta Sufaru, Maria Manaila, Ingrid Ioana Stafie, Gabriela Melinte, Bianca Simionescu, and Letitia Leustean. 2022. "Pathogenic Connections in Post-COVID Conditions: What Do We Know in the Large Unknown? A Narrative Review" Viruses 14, no. 8: 1686. https://doi.org/10.3390/v14081686
APA StyleStafie, C. S., Solomon, S. M., Sufaru, I.-G., Manaila, M., Stafie, I. I., Melinte, G., Simionescu, B., & Leustean, L. (2022). Pathogenic Connections in Post-COVID Conditions: What Do We Know in the Large Unknown? A Narrative Review. Viruses, 14(8), 1686. https://doi.org/10.3390/v14081686