1. Introduction
Nipah virus (NiV) is a zoonotic emergent virus of the
Paramyxoviridae family, with a fatality rate reaching 92% in humans. This virus represents a threat for public health and can cause considerable economic repercussions, since it can also infect pigs [
1]. Among NiV cytopathogenic mechanisms, its ability to rapidly form syncytia is a hallmark of infection. As with other members of the
Mononegavirales, the accumulation of inclusion bodies (IB) in the cytoplasm of NiV-infected cells is a very common phenomenon [
2,
3,
4,
5]. Such IB are generally described as an aggregation of viral proteins that in some cases, can function as viral factories inside the cell [
6].
Interestingly, the accumulation of certain proteins with a specific amino acid composition and disposition facilitates nucleation, thereby, in some cases, inducing their precipitation as crystals. In fact, it has been observed that such protein-derived aggregates can naturally appear in living cells; for instance, insulin crystals have been shown to form in secretory granules [
7]. This process is known as in cellulo, or in vivo, crystallization and can be classified as a native or disease associated intracellular process, which can be harmless, harmful, or useful for the cell [
8,
9]. As a primary function, native in cellulo aggregation supports protein storage (e.g., as a nutrient supply), and encapsulation has also been described as a primary function of some in cellulo crystals [
10,
11]. Other functions associated with the in cellulo formation of ordered aggregates or crystal-like structures include compartmentalization [
12], wound sealing [
13], and the removal of toxins. However, disease-associated crystallization is believed to be a defense mechanism in response to stress or cellular damage [
14,
15].
While highly ordered structures may be easily observed with basic optical microscopy, super-resolution microscopy techniques, electron microscopy, or diffraction by X-ray crystallography are commonly used to define their crystalline state by visualization of the crystal lattice or detection of crystal-specific Bragg diffraction [
16,
17].
Ordered intracellular structures or crystals can assume many different shapes (e.g., rhombohedral, rhomb-dodecahedral, bipyramidal, hexagonal, or rectangular) and occur with no apparent preference for a specific cellular compartment, being frequently present in the cellular cytosol but also in the endoplasmic reticulum (ER), peroxisomes, lysosomes, nucleus, and mitochondria, as summarized by Mudogo et al. [
18]. However, individual crystal-like aggregates of a specific protein usually have a growth preference for a specific compartment [
19].
While in recent years, an increasing number of publications have reported in cellulo crystallization/ordered aggregation, the specific physicochemical parameters and molecular mechanisms required to generate such structures in cells have not yet been fully elucidated, and in many cases, the formation of these structures remains unperceived. In this study, we describe the presence of intracellular, rhombohedron-shaped ordered clusters during NiV infection in Vero E6 and Huh-7.5 cells. Although a typical crystal lattice is not observed internally by electron microscopy, these structures present external characteristic sharp edges, which is an indication of ordered structure. Furthermore, we provide a description of their appearance during infection, their formation kinetics, and their subcellular localization.
2. Materials and Methods
2.1. Cells and Infections
Vero E6 (ATCC, Vero C1008, clone E6, Manassas, VA, USA) and Huh-7.5 (Lonza, Cologne, Germany) cells were cultivated in Dulbecco’s modified Eagle’s medium (DMEM, PanBiotech, Aidenbach, Germany) supplemented with 100 U/mL of Pen/Strep (Gibco, Thermo Fisher Scientific, Paisley, UK) and 5% or 10% of fetal bovine serum (FBS, Gibco, Thermo Fisher Scientific), respectively.
All experiments were performed with NiV (Malaysia strain, accession number MK673562.1), EBOV (Mayinga strain, accession number KR063671.1), and JUNV (Junín XJ strain, accession number JF799981.1 and JF799977.1), under BSL-4 conditions, in the Bernhard Nocht Institute for Tropical Medicine (Hamburg, Germany). All infections were conducted at an MOI of 0.1. Briefly, 5 × 104 Vero E6 or Huh-7.5 were seeded on glass coverslips in 24-well plates, and the following day, were infected for one hour at 37 °C, before removal of the inoculum and addition of DMEM supplemented with 2.5% of FCS. After the specified infection times, cells were fixed with either 4.5% formaldehyde solution (SAV Liquid Production GmbH, Flintsbach am Inn, Germany) or a 1:1 dilution of acetone:methanol (Ac:MeOH) (Sigma-Aldrich, Burlington, MA, USA and Carl Roth, Karlsruhe, Germany respectively) for one hour at room temperature (RT).
2.2. Immunofluorescence Analysis
A total of 5 × 10
4 Vero E6 and Huh-7.5 were seeded onto glass coverslips (number 1, 12 mm, assistant, Sondheim vor der Rhön, Germany) in 24-well plates and infected at 70% confluency with the respective viruses. At various time points post-infection (p.i.), namely 8 h p.i., 18 h p.i., 24 h p.i., or 48 h p.i., infected cells were inactivated with either 4.5% formaldehyde solution or a 1:1 dilution of Ac:MeOH for 1 h at RT. Paraformaldehyde (PFA) inactivated samples were washed with PBS (PanBiotech, Aidenbach, Germany) 3 times for 5 min, prior to being permeabilized in 0.1% (
v/
v) Triton-X-100 (Carl Roth) in PBS for 10 min at RT. The Ac:MeOH inactivated samples did not require such permeabilization and were air-dried in a chemical hood. After additional washing steps, as described above, 1% (
w/
v) bovine serum albumin (BSA, Sigma Aldrich, Darmstadt, Germany) in PBS was added to the cells to block unspecific binding for 30 min at RT. After washing, cells were stained with the respective primary antibodies, diluted in PBS for 1 h at RT in a wet chamber. The primary antibodies used were NiV N antibody (1:750, mouse, kindly provided by Indre Kucinskaite-Kodze from the Life Science Center at Vilnius University, Vilnius, Lithuania), JUNV NP antibody (1:300 mouse, BEI Resources, Clone IC06-BE10), in-house mouse polyclonal anti–pan-Ebolavirus NP primary antibody (1:2000, 1 h at RT), anti-human Golgi Glg1 (1:50, rabbit IgG, Invitrogen—Thermo Fisher Scientific, Waltham, MA, USA, PA5-26838), and anti-human peroxisome Pmp70 (1:100, rabbit IgG, Invitrogen, PA1-650). Following extensive washing, secondary antibodies goat anti mouse FITC (1:100, Thermo Fisher Scientific, 62-6511), goat anti mouse AF647 (1:1000, Thermo Fisher Scientific, A32728), or donkey anti rabbit AF647 (Thermo Fisher Scientific, A-31573), diluted in PBS, were incubated for 1 h at room temperature in a dark, wet chamber. If applicable, actin (Phalloidin, 1X, Sigma, P5282) or microtubule (Flutax-2, 1 µM, Thermo Fisher Scientific, P22310) staining was performed during this incubation period. Upon completion of the final washing steps, cover slips were dipped into ddH
2O, dried, and mounted onto microscopy slides using Glycerol Mounting Medium with DAPI and DABCO
TM (Abcam, Cambridge, UK) for counterstaining of the nuclei. Imaging was performed with Zeiss Axio Imager M1 microscopes, or a Leica SP8 confocal microscope. For image analysis, FIJI with Bio-Formats plug-in for Mac OS X was employed [
20].
2.3. Quantification of NiV Ordered Structures Size
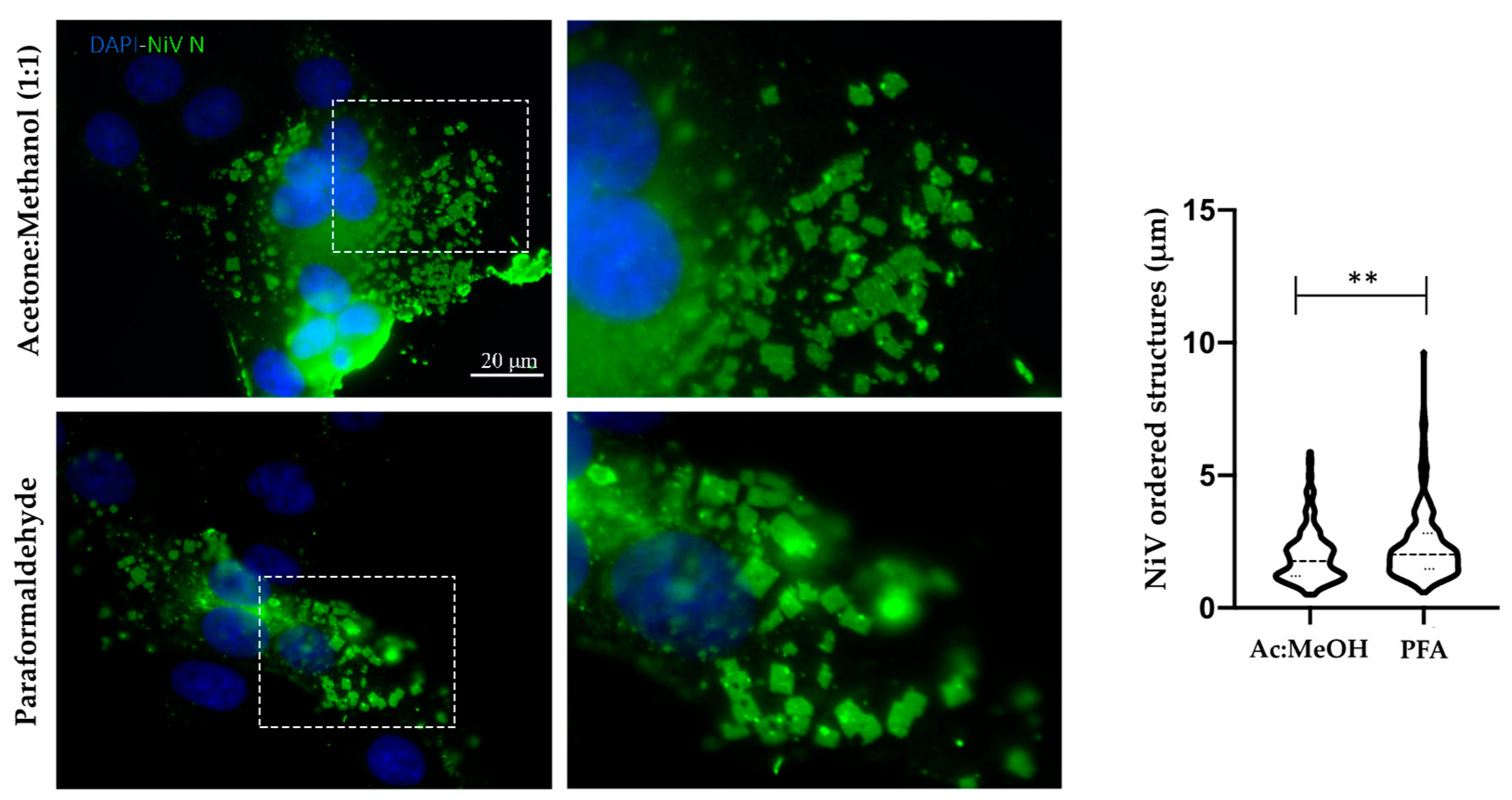
Sizes of the organized structures observed for NiV were estimated by using standard tools and plugins provided in ImageJ (v.1.51) software. Briefly, the Straight Line tool in FIJI was used to define the length of one side of NiV ordered structures and subsequently, these lines were added as regions of interest (ROIs) to the ROI Manager. Once one side of each ordered structure was traced, its length was obtained by using the command “measure” and choosing “shape descriptors” as output. The length of one side per ordered structure was determined in 9 fields for Ac:MeOH fixation and PFA fixated samples, respectively, in three independent experiments. Size distribution was then analyzed with GraphPad Prism version 8.0.1 for Windows (San Diego, CA, USA).
2.4. Super-Resolution Microscopy, Structural Illumination Microscopy
For super-resolution microscopy (SRM) via structural illumination microscopy (SIM), Huh-7.5 cells grown on cover slips (#1.5, 10 mm, Carl Roth, Karlsruhe, Germany) were infected at an MOI of 0.1 and at 18 h p.i., cells were fixed with 4.5% formaldehyde, as described above. The cells were stained according to the protocol detailed above, using NiV N primary antibody (1:500 dilution), followed by a secondary goat anti mouse antibody conjugated to FITC (1:100, Thermo Fisher Scientific, 62-6511). Fluorescence Mounting Medium (Dako, Agilent Technologies, Santa Clara, CA, USA) was used to mount the cover slips onto microscopy slides. Imaging was performed with a Nikon-N-SIM microscope with a 488 nm laser and an apochromatic 100× oil immersion objective (NA 1.49).
2.5. Transmission Electron Microscopy (TEM)
A total of 5 × 105, 7.5 × 105 or 1.25 × 106 Vero E6 cells were plated in a high Grid-500 35 mm μ-Dish (IBIDI). Cells were infected at an MOI of 0.1, and after 24 h, cells were fixed for 1 h in 4.5% PFA and postfixed with 2.5% glutaraldehyde (GA) overnight. Subsequently, cells were washed with PBS, postfixed for 30 min with 1% OsO4 in PBS, washed with ddH2O, and stained with 1% uranyl acetate in water. The samples were gradually dehydrated with ethanol and embedded in Epon resin (Carl Roth, Karlsruhe, Germany) for sectioning. Ultrathin 50 nm sections were prepared using an Ultracut Microtome (Leica Microsystems, Weitzlar, Germany). The sections were poststained with 1% uranyl acetate. Electron micrographs were obtained with a 2 K wide angle CCD camera (Veleta, Olympus Soft Imaging Solutions GmbH, Münster, Germany) attached to a FEI Tecnai G 20 Twin transmission electron microscope (FEI, Eindhoven, The Netherlands) at 80 kV.
2.6. High Resolution Scanning Electron Microscopy (SEM)
After 24 h p.i., NiV-infected Vero E6 cells were fixed with 4.5% formaldehyde and were permeabilized in 0.1% Triton-X-100 in PBS for 10 min at RT, stained according to the protocol detailed above, using NiV N primary antibody (1:500 dilution), followed by a secondary goat anti-mouse antibody conjugated to FITC. Samples were processed and analyzed for TEM, as described above. For SEM, samples were dehydrated in a graded series of ethanol, air-dried after transition to pentane, and finally sputtered with 5 nm gold. Images were obtained in a Tescan Clara scanning microscope operated at 3 kV, using an ET secondary electron detector.
2.7. Correlative-Light Electron Microscopy (CLEM)
A total of 5 × 104 Vero E6 cells were seeded in a high Grid-500 35 mm μ-Dish (IBIDI) and were infected with NiV at an MOI of 0.1 24 h later. At 24 h post-infection, the dishes containing NiV infected cells were fixed for 1 h in 4.5% formaldehyde. The inactivated samples were washed with PBS 3 times for 5 min, prior to being permeabilized in 0.1% (v/v) Triton-X-100 in PBS for 10 min at RT. Then, cells were blocked with 1% BSA in PBS for 30 min at RT. After washing, cells were stained with NiV N antibody for 1 h at room temperature in a wet chamber. Following extensive washing, secondary antibodies goat anti mouse FITC (1:100) were incubated for 1 h at room temperature in a dark, wet chamber. Fluorescence imaging was carried out with a spinning disc microscope (Nikon Ti2E equipped with a Yokogawa CSU-W1 spinning disc unit, an Andor iXon Ultra EMCCD camera, and a 100× NA 1.49 plan apo oil immersion objective. The samples were gradually dehydrated with ethanol and embedded in Epon resin (Carl Roth, Germany) for sectioning. Imaging was performed using a FEI Tecnai G 20 Twin transmission electron microscope. The samples were also processed for TEM, as described above. Electron micrographs were obtained with a 2K wide angle CCD camera (Veleta, Olympus Soft Imaging Solutions GmbH, Münster, Germany) attached to a FEI Tecnai G 20 Twin transmission electron microscope (FEI, Eindhoven, The Netherlands) at 80 kV.
2.8. Protein In Silico Analysis
The amino acid sequences of NiV (AAX51852.1), EBOV (AAD14590.1), and JUNV (ACS12871.1) nucleoproteins and NiV matrix protein (NP_112025.1) were analyzed with ProtParam (
https://web.expasy.org/protparam/, accessed on 8 December 2021) software from Expasy to determine the percentage of hydrophobic amino acids contained in their sequences [
21]. In order to estimate their solubility index, Protein-Sol (
https://protein-sol.manchester.ac.uk, accessed on 8 December 2021) software from Expasy was used [
22].
The CamSol method from The Chemistry of Health Software (
https://www-cohsoftware.ch.cam.ac.uk/index.php/camsolintrinsic, accessed on 29 December 2021) was used for the calculation of the intrinsic solubility profile and the score of each analyzed protein [
22]. Monomeric 3D model structures of NiV N, NiV P, and NiV M were calculated with Alphafold software (
https://alphafold.ebi.ac.uk, accessed on 8 December 2021, [
23,
24]) using the associated protein sequences with the accession numbers AY858110.1, HM545087.1, and NP_112025.1, respectively. The Swiss-Model server (
https://swissmodel.expasy.org/interactive, accessed on 8 December 2021) was used for modeling of the 3D structures of the multimeric forms of NiV N, NiV P, and NiV M. Among the 8 templates available for NiV N in the Protein Data Bank (PDB;
www.rcsb.org, accessed on 8 December 2021), the selected template, PDB 7NT5 obtained by cryoEM at 3.5 Å resolution, showed an identity of 98.68%, coverage of 1.00, QMEAN: 0.81 ± 0.05, global model quality estimation (GMQE) of 0.76, and quaternary structure quality estimation (QSQE) of 0.78. For structural modeling of NiV P, 28 templates are available in the PDB. The selected template 4GJW was obtained by X-ray crystallography at 3.0 Å resolution; it showed an identity of 99.08%, coverage of 0.15, QMEAN: 0.72 ± 0.05, GMQE: 0.06, and QSQE: 0.40. Among the 27 templates available for NiV M, the selected sample 6BK6 was obtained with X-ray crystallography at 2.5Å resolution, showing an identity of 91.81%, coverage of 0.97, QMEAN: 0.86 ± 0.05, GMQE: 0.84, and QSQE: 0.82. Pymol software was used to generate 3D protein structure images and videos [
25].
4. Discussion
Despite the intracellular formation of well-ordered assemblies, including protein crystals, being a known phenomenon for more than a century [
33], this represents a relatively unknown process that frequently occurs unnoticed. The presence of ordered or crystal-like structures may be observed in living cells, but they are not recognized as such. This may be the case for NiV infected cells, in which two different “inclusion body” types have been described to date: perinuclear IB (IB
peri) and plasma membrane IB (IB
pm) [
28]. Based on our findings, we believe that what were previously referred to as IB
pm, are not, in fact, inclusion bodies, but distinct, rhombohedron-shaped NiV-ordered viral structures.
In order to visualize biological samples in detail through microscopy, it is necessary to fix them. The fixation process used is crucial, since it determines the level of resemblance between the image seen in the microscope and the in vivo structure. Thus, it is important to avoid the generation of artifacts due to sample manipulation. There are several ways to carry out chemical fixation, but some of the most commonly used methods for microscopy include the use of aldehydes and alcohols. Aldehydes are additive fixation solutions that generate covalent interactions (cross-links) between proteins and preserve their natural structure. In comparison with other aldehydes, such as glutaraldehyde, paraformaldehyde is known to have less effect in terms of the masking of a protein’s antigenicity during fixation; therefore, it is commonly used [
26]. On the other hand, alcohols are denaturing or precipitating fixators which can denature the protein when they reduce their solubility, thus modifying their tertiary structure; alcohols are frequently used in combination with other chemicals such as acetone in order to improve the fixation process [
26,
34].
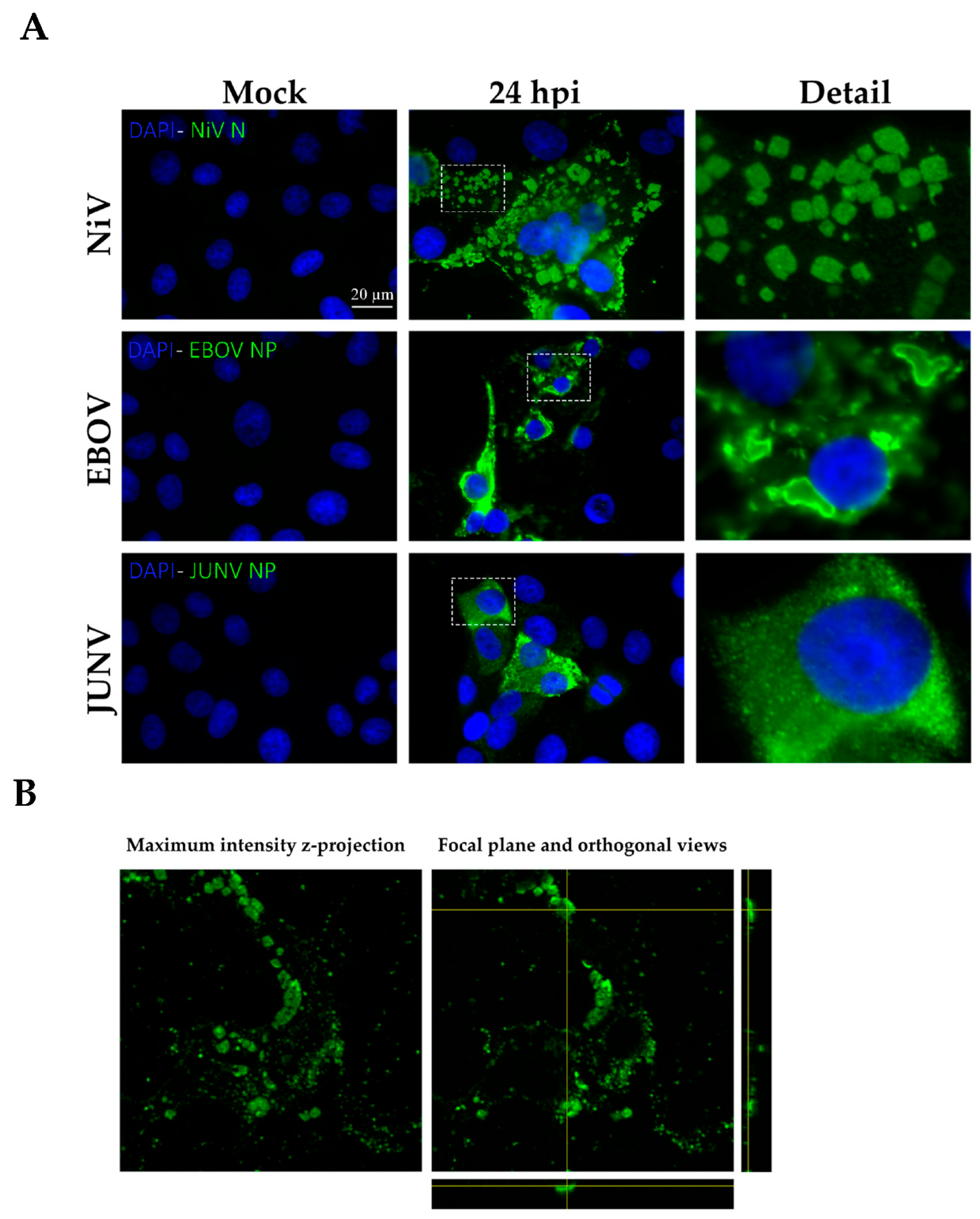
In our study we compared both types of chemical fixation, additive and denaturing, in order to eliminate the possibility that the formation of NiV-ordered structures is artificially associated with the fixation process. Moreover, we fixed cells infected by other viruses, one from the same Mononegavirales order (EBOV) and one from Bunyavirales (JUNV), using both fixation methods in order to determine the specificity of ordered structure formation during NiV infection. We observed that these structures specifically occur in NiV-infected cells, independent of the fixation method.
Several factors, including cellular elements, may contribute to this process during NiV infection; while it is not the only contributing intrinsic factor, the presence of hydrophobic amino acids, particularly at the surface of the viral protein structures, may facilitate the unique, ordered aggregation of the NiV N and M proteins [
35,
36]. This is in line with a previous study that confirmed the presence of NiV N and M in comparable ordered structures detected after NiV infection, even if they have been classified as IBs [
28]. Consistent with less exposure of hydrophobic patches and the increased predicted solubility, no indications of the presence of NiV P were found in these structures.
While the intrinsic solubility of a protein is relevant to determine its aggregation tendency, other factors such as the specific cellular physicochemical environment, the content, polarity and disposition of the amino acids, the levels of gene expression, and the viral complexity itself play a major role in this process. It has been shown that intracellular aggregation first requires a high local concentration of the protein, along with precipitant agents, in order to render aggregation thermodynamically favorable, thus reaching the required protein supersaturation [
18]. NiV N and M proteins are abundantly produced during NiV replication, and importantly, we have shown that an increase in NiV N over time coincides with the formation of the specific NiV N-ordered structures. However, it has been reported that the NiV matrix protein M is additionally required to form comparable ordered structures, previously described as IB
pm [
28]. Indeed, it has been shown by others that the expression of NiV N protein alone or in combination with part of the NiV P protein does not induce the formation of characteristic structures in transfected cells [
37]. Thus, it seems that both NiV N and NiV M are involved in or affected by this aggregation process.
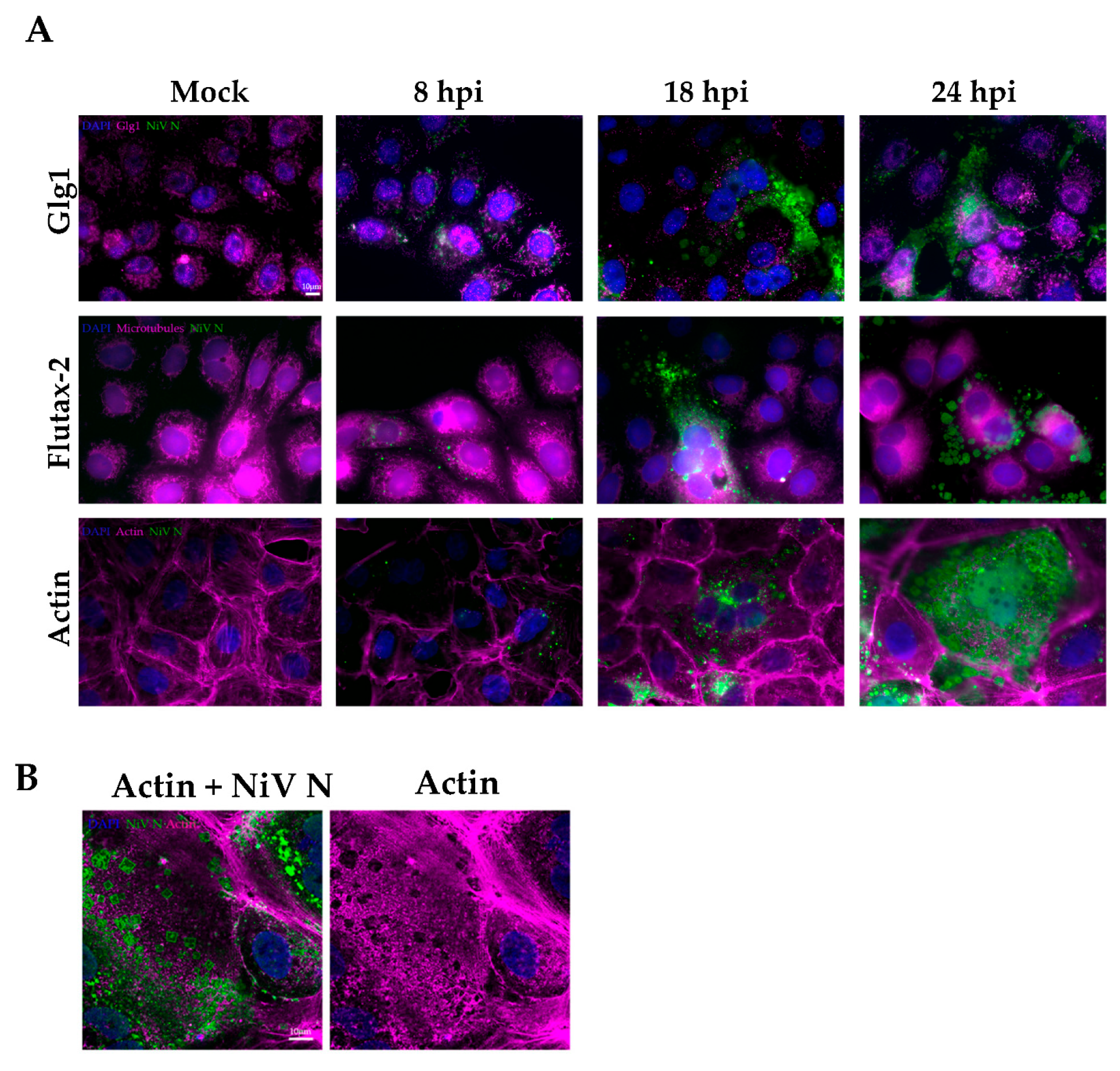
The localization of intracellular aggregates in cells can be very variable, as they can appear either in cellular organelles or in the cytoplasm. NiV ordered structures were found to be exclusively located in the cytoplasmic compartment of the cell, particularly in the periphery of infected cells, or associated with syncytia. Whether they form there for strategic functional reasons, or are simply placed there by physiological forces remains to be determined. While it has been suggested that analogous NiV structures (described as IB
pm) may have a role related to virion assembly and budding, it has not been proven that fusion events take place between IB
pm and the cellular membrane [
28]. In contrast to this theory, our findings suggest that NiV-ordered structures may displace the actin cytoskeleton, which usually plays an important role during membrane fusion and viral syncytia formation [
38,
39]. The involvement of actin in syncytia formation and viral release has previously been shown for orthomyxoviruses (namely influenza) and other paramyxoviruses members, such as respiratory syncytial virus (RSV) [
40,
41]. Importantly, our observation of NiV-ordered structures of up to 10 µm in size suggests that a role related to multiplicity and the formation of inclusion bodies, as suggested in previous studies [
28], is dubious. However, the fact that regular NiV clusters are observed by TEM and SEM exclusively in the periphery of the cellular membrane does not negate the possibility that these structures are related to the budding processes. Indeed, the fact that NiV-organized structures have a rhombohedral form that can be disrupted during budding provides a new insight of how these structures act during NiV infection.
This study provides a detailed physical characterization of NiV-organized structures that could help to further investigate their role, which is likely of functional importance. In addition, the aggregation of viral proteins may be a virulence factor, since this phenomenon concentrates viral proteins over time, but could also represent a defensive mechanism of the virus against proteolytic degradation. While it is suspected that such an abnormal protein assembly mechanism conveys a viral strategy which may have been evolutionary optimized, an accidental event cannot be excluded either. In fact, NiV protein aggregation may well be a defense mechanism of the cell, preventing cell cytotoxicity, or alternatively, have a more specific function related to viral infection, transmission, or replication. While it is possible that crystal formation could be advantageous for cells in some cases, the high concentration and size of NiV in cellulo-ordered structures may be detrimental to the functions of the cell. Consequently, it would be interesting to observe if such structures are formed in vivo.
The presented data confirms a specific degree of order in the NiV clusters, but does not allow for confirmation of the crystalline character of the detected structures. Indeed, we were unable to clearly visualize a well-ordered lattice structure by TEM, or to detect specific Bragg diffraction of X-rays at the P14 beamline using the synchrotron source PETRA III (German Electron Synchrotron DESY, Hamburg, Germany; data not shown), which are both accepted as a proof for crystallinity. Despite the high brilliance of the X-rays provided, the latter might be limited by the extremely small diffractive volume of the structures [
16]. However, the characteristic rhombohedral shape of the distinct structures, combined with the frequently observed and the extremely sharp edges, strongly indicate a high degree of order.
Moreover, the ordered structures appear to be virus-specific and are not formed as artifacts of the fixation process, supported by their association with high aggregates of the NiV nucleoprotein and the numerous RNC-like structures seen with electron microcopy. The specific location in the cellular periphery suggests that this preferred localization might be due to physiological constraints. Further studies involving in vitro and in vivo analysis techniques are required to understand the molecular features promoting and regulating the formation of intracellular ordered structures during NiV infection. Our data highlights the frequency with which NiV-ordered structures appear during infection and emphasizes the importance of investigating their role in more detail, thus providing a potential target for novel therapeutic directions, with the aim of preventing their formation or disrupting these potentially detrimental complexes during NiV infection.


 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







