
Flavivirus Capsid Proteins Inhibit the Interferon Response

 , , and
, , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Mammalian Cell Culture and Virus Infections
2.2. Production of Lentivirus and Transduction of Cells
2.3. Sample Preparation for RNA-Sequencing
2.4. Ion Torrent Next-Generation Sequencing
2.5. Differential Gene Expression (DEG) Analysis
2.6. RNA Extraction and qRT-PCR
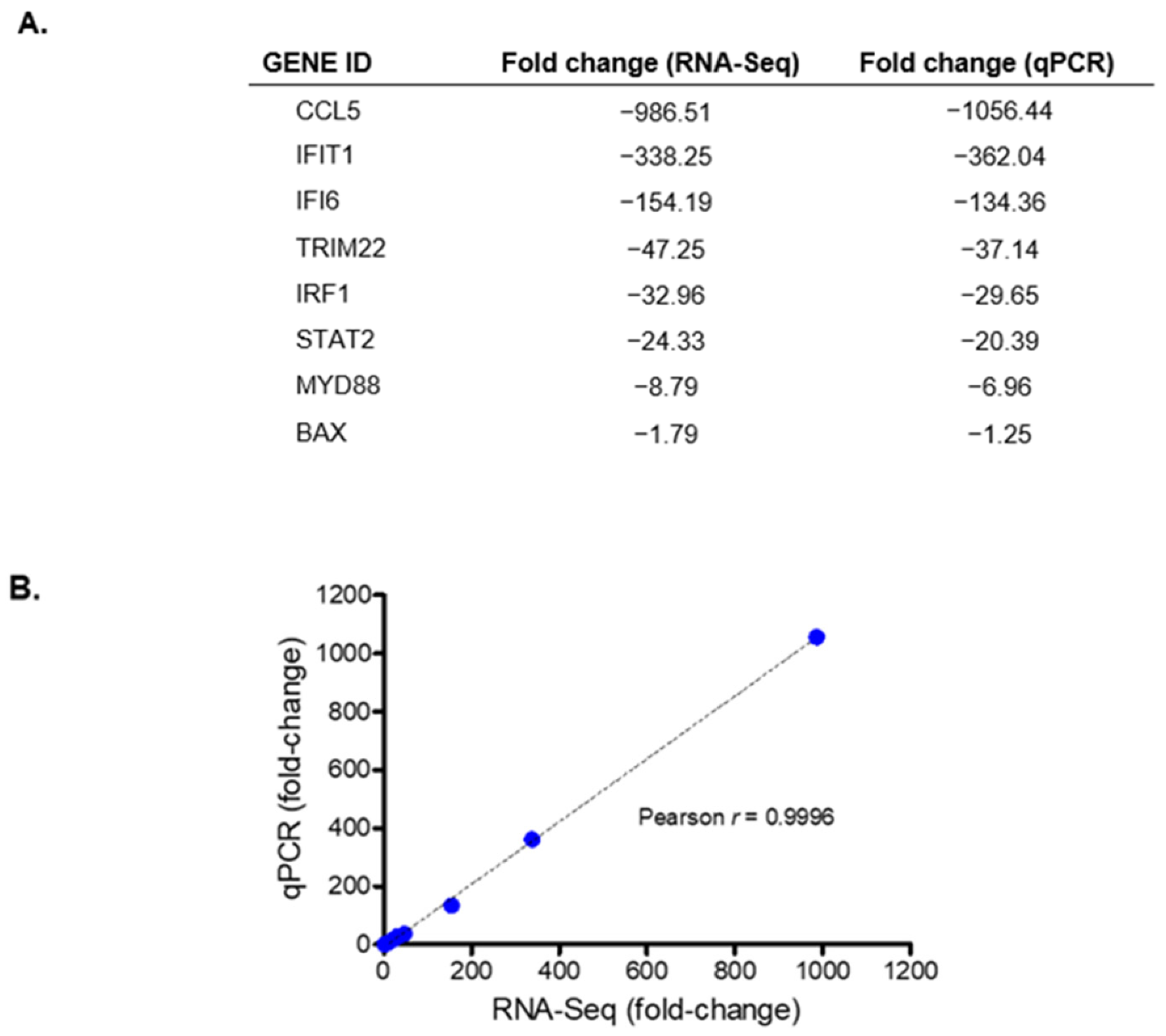
2.7. Validation of RNA-Seq
2.8. Immunoblotting
2.9. Antibodies
2.10. Flow Cytometry Analysis of ZIKV-Infected Cells
2.11. Generation of Stably Transduced Cell Lines
2.12. Polyinosinic:Polycytidylic Acid (Poly[I:C]) Stimulation
2.13. Affinity Purification Mass Spectrometry (AP-MS)
2.14. Co-Immunoprecipitation
2.15. Knockdown of TRIM25
2.16. Over-Expression of TRIM25
2.17. GST Pulldown and Immunoblotting
3. Results
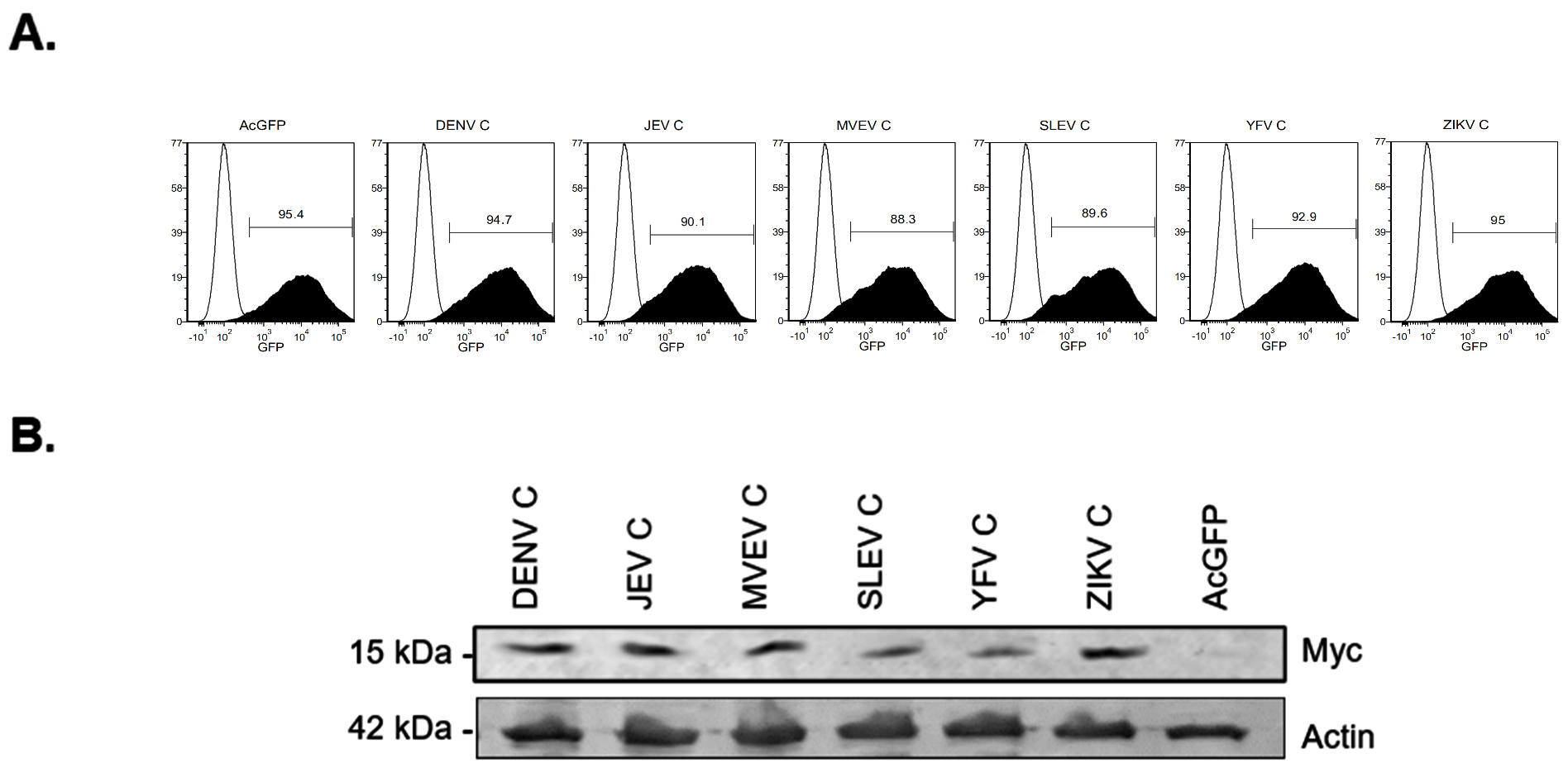
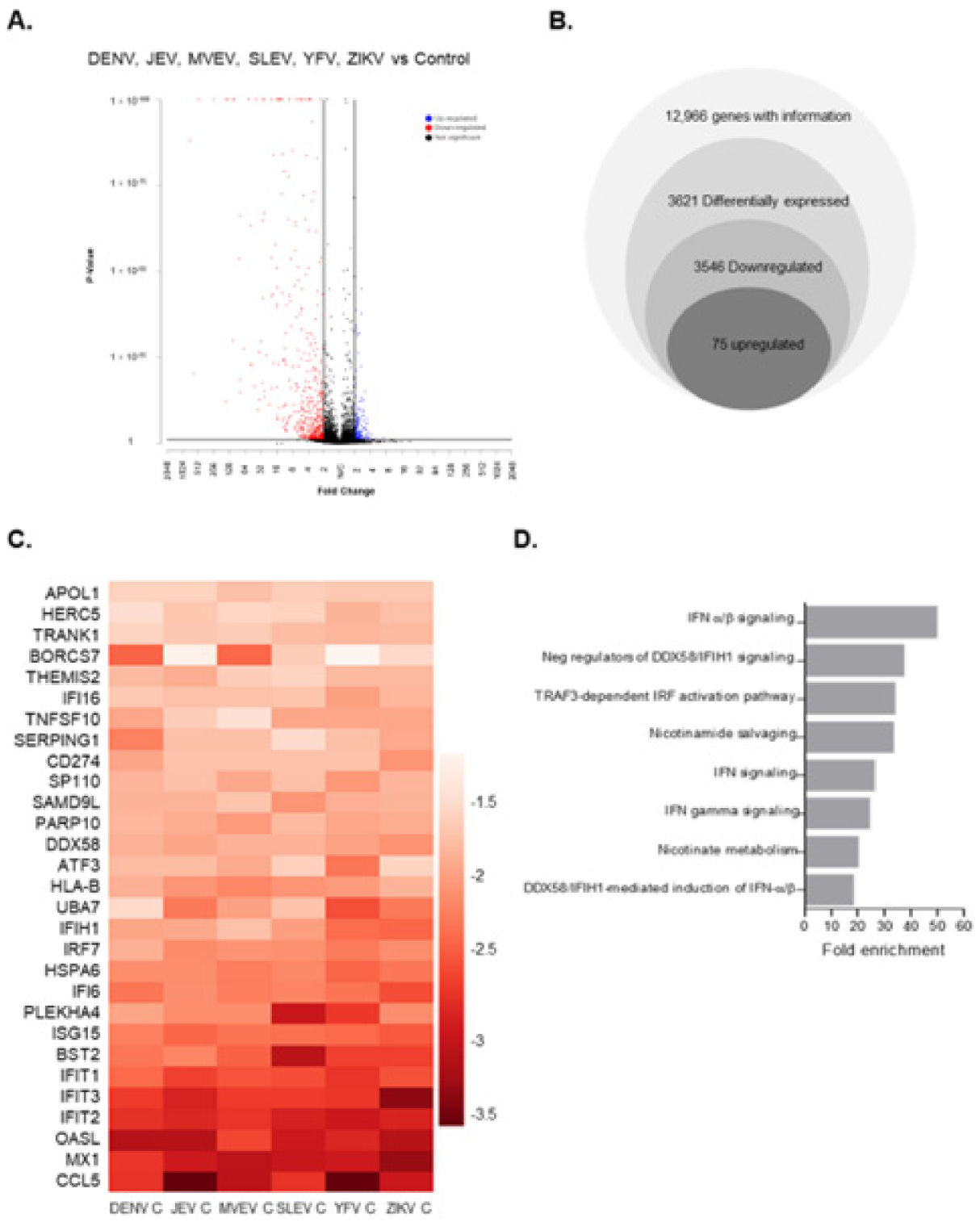
3.1. Global Gene Expression Changes in Capsid-Expressing Cells
3.2. Validation of Transcriptomic Changes Observed in RNA-Seq Analyses
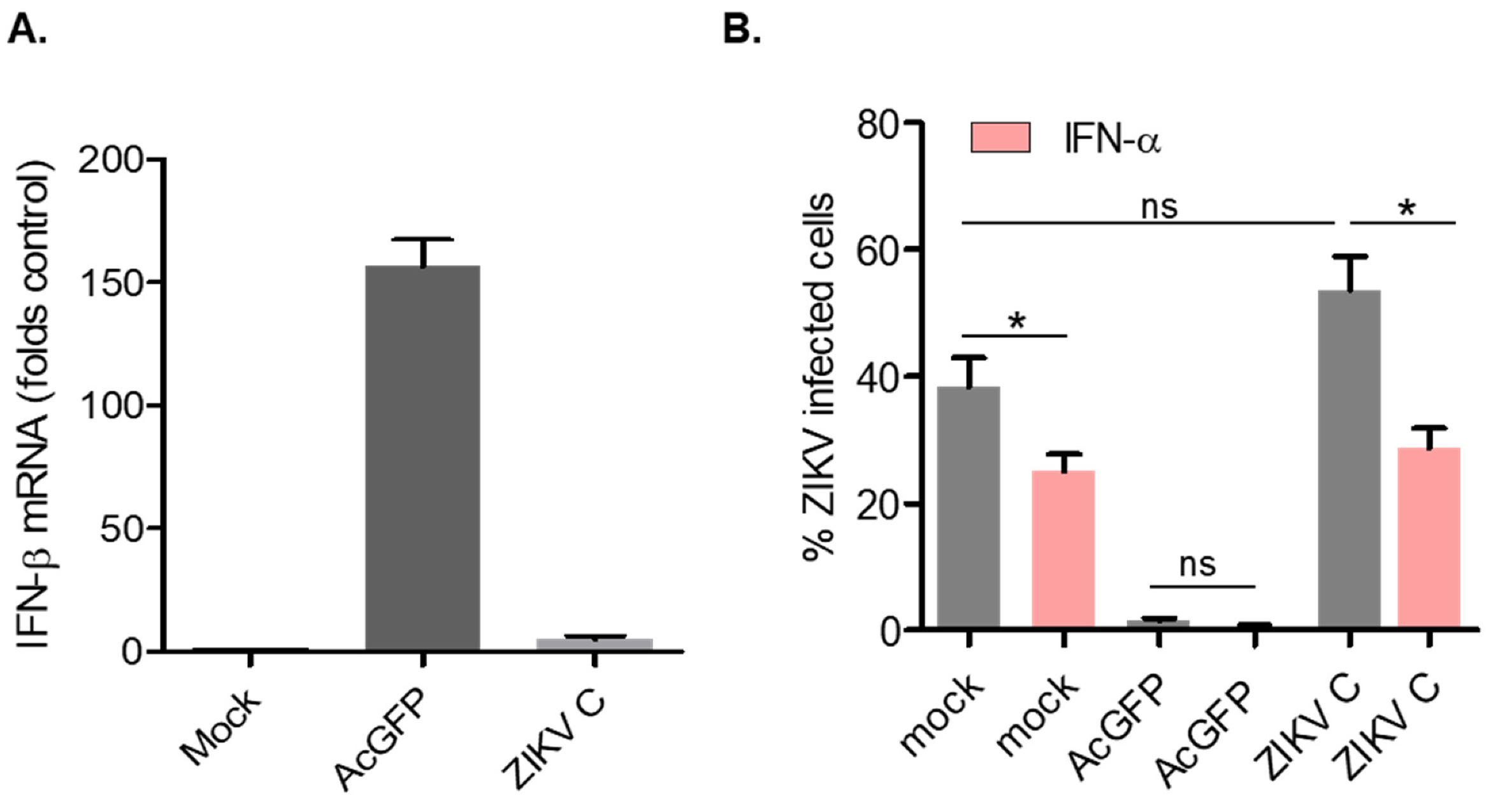
3.3. Cells Transduced with Lentiviruses Encoding ZIKV Capsid Suppress IFN-β and Are Susceptible to Viral Infection
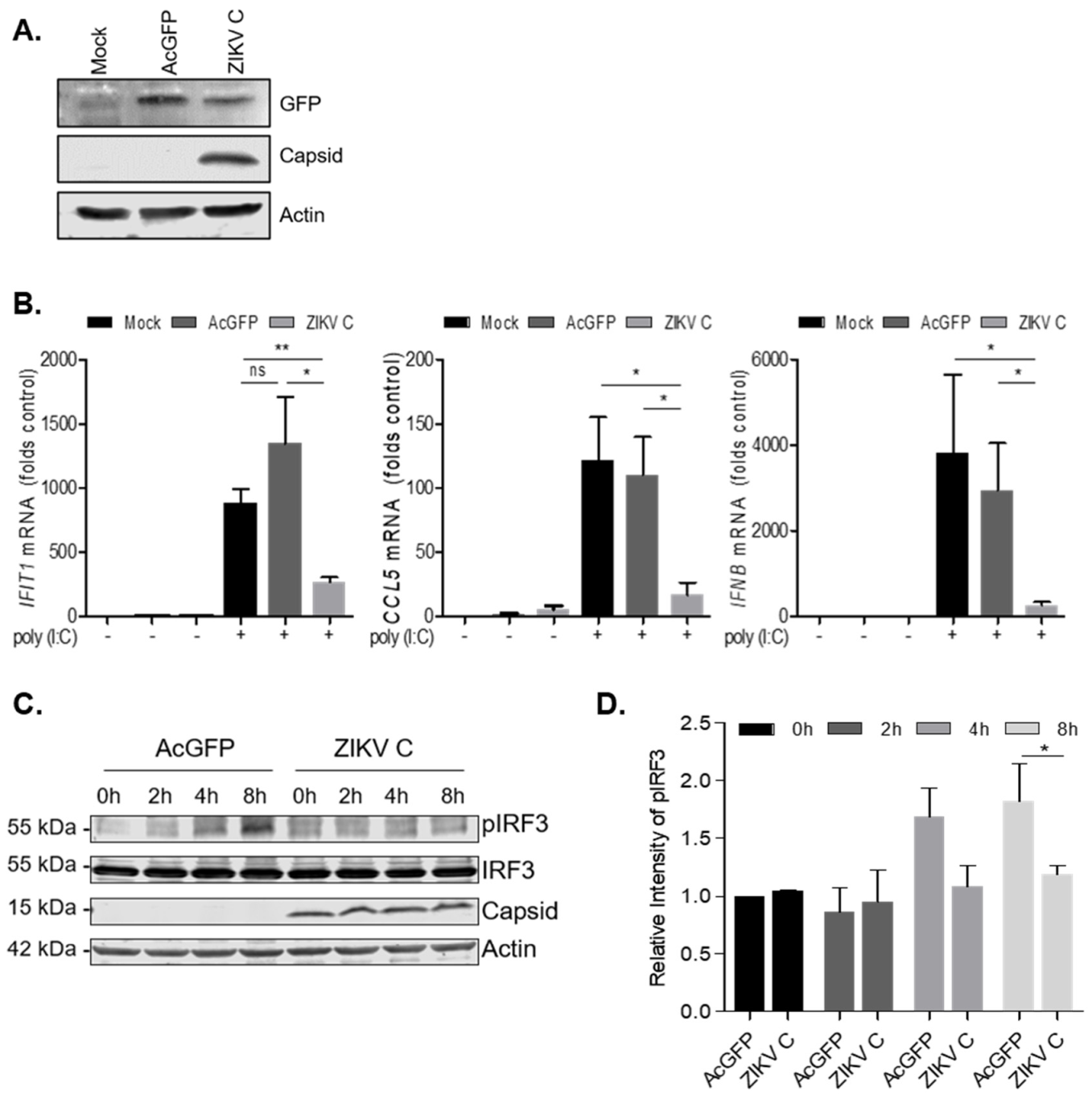
3.4. ZIKV Capsid-Expressing Cells Subvert IFN Induction
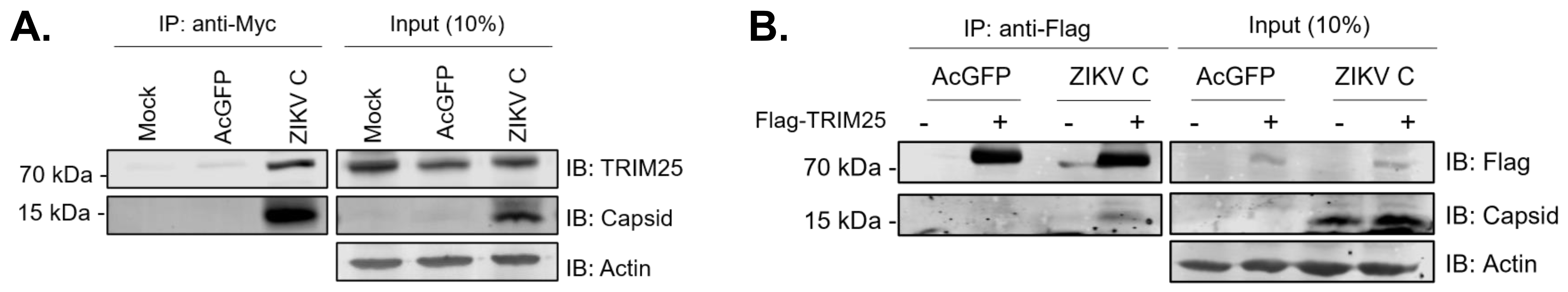
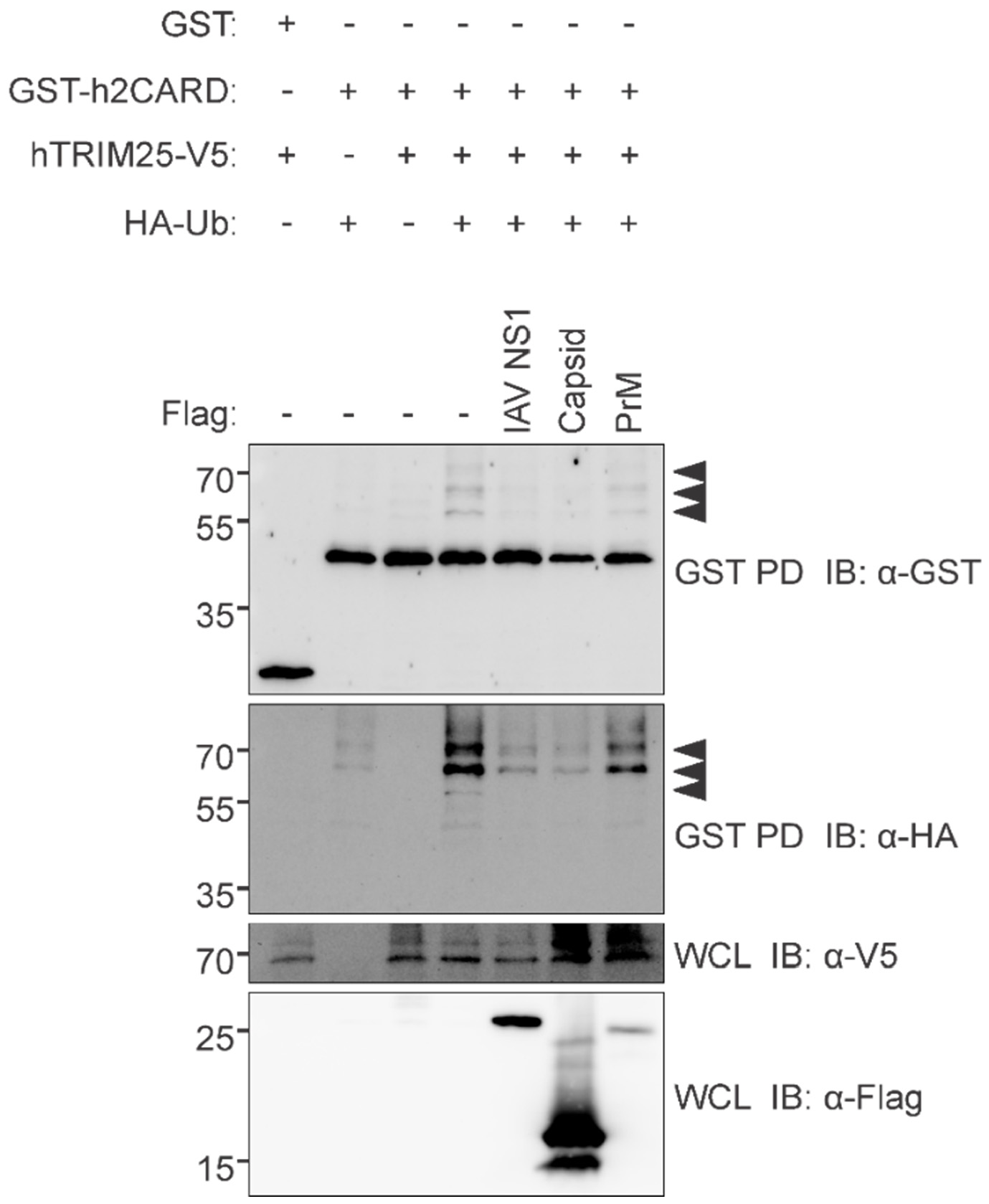
3.5. ZIKV Capsid Interacts with TRIM25 and Suppresses RIG-I Ubiquitination
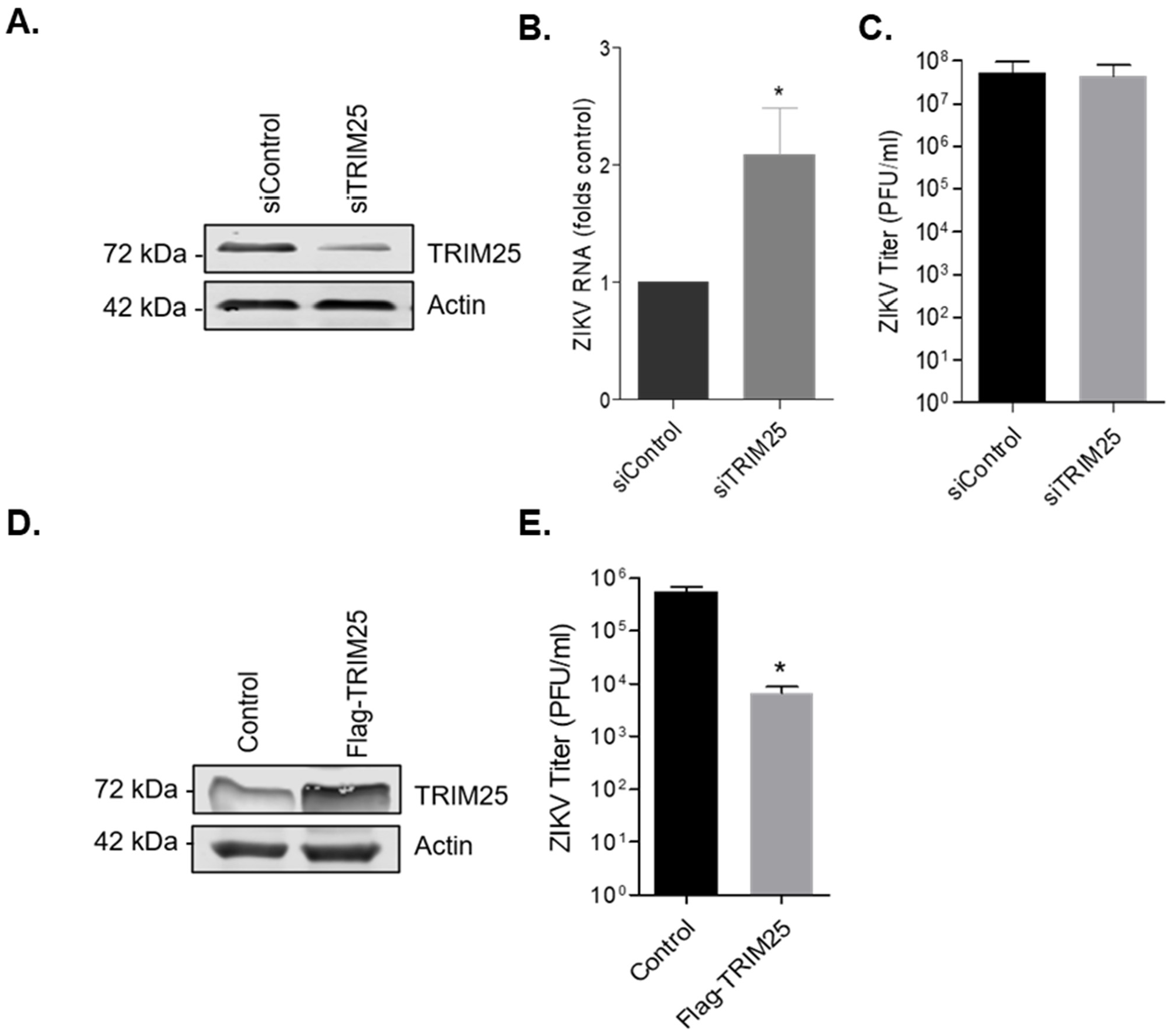
3.6. TRIM25 Is a Viral Restriction Factor
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dick, G.; Kitchen, S.; Haddow, A. Zika virus (I). Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- Gubler, D.J.; Vasilakis, N.; Musso, D. History and emergence of Zika virus. J. Infect. Dis. 2017, 216 (Suppl. S10), S860–S867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffy, M.R.; Chen, T.-H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C. Zika virus outbreak on Yap Island, federated states of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Cao-Lormeau, V.-M.; Roche, C.; Teissier, A.; Robin, E.; Berry, A.-L.; Mallet, H.-P.; Sall, A.A.; Musso, D. Zika virus, French polynesia, South pacific, 2013. Emerg. Infect. Dis. 2014, 20, 1085. [Google Scholar] [CrossRef]
- Faria, N.R.; Quick, J.; Claro, I.; Theze, J.; de Jesus, J.G.; Giovanetti, M.; Kraemer, M.U.; Hill, S.C.; Black, A.; da Costa, A.C. Establishment and cryptic transmission of Zika virus in Brazil and the Americas. Nature 2017, 546, 406. [Google Scholar] [CrossRef]
- Faria, N.R.; da Silva Azevedo, R.d.S.; Kraemer, M.U.; Souza, R.; Cunha, M.S.; Hill, S.C.; Thézé, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.K.; Shin, O.S. Advances in Zika virus–host cell interaction: Current knowledge and future perspectives. Int. J. Mol. Sci. 2019, 20, 1101. [Google Scholar] [CrossRef] [Green Version]
- Krauer, F.; Riesen, M.; Reveiz, L.; Oladapo, O.T.; Martinez-Vega, R.; Porgo, T.V.; Haefliger, A.; Broutet, N.J.; Low, N.; Group, W.Z.C.W. Zika virus infection as a cause of congenital brain abnormalities and Guillain–Barré syndrome: Systematic review. PLoS Med. 2017, 14, e1002203. [Google Scholar] [CrossRef] [Green Version]
- Morrison, T.E.; Diamond, M.S. Animal models of Zika virus infection, pathogenesis, and immunity. J. Virol. 2017, 91, e00009-17. [Google Scholar] [CrossRef] [Green Version]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A mouse model of Zika virus pathogenesis. Cell Host. Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef] [Green Version]
- Samuel, M.A.; Diamond, M.S. Alpha/beta interferon protects against lethal West Nile virus infection by restricting cellular tropism and enhancing neuronal survival. J. Virol. 2005, 79, 13350–13361. [Google Scholar] [CrossRef] [Green Version]
- Shresta, S.; Kyle, J.L.; Snider, H.M.; Basavapatna, M.; Beatty, P.R.; Harris, E. Interferon-dependent immunity is essential for resistance to primary dengue virus infection in mice, whereas T-and B-cell-dependent immunity are less critical. J. Virol. 2004, 78, 2701–2710. [Google Scholar] [CrossRef] [Green Version]
- Johnson, A.J.; Roehrig, J.T. New mouse model for dengue virus vaccine testing. J. Virol. 1999, 73, 783–786. [Google Scholar] [CrossRef] [Green Version]
- Meier, K.C.; Gardner, C.L.; Khoretonenko, M.V.; Klimstra, W.B.; Ryman, K.D. A mouse model for studying viscerotropic disease caused by yellow fever virus infection. PLoS Pathog. 2009, 5, e1000614. [Google Scholar] [CrossRef] [Green Version]
- Thompson, M.; Kaminski, J.; Kurt-Jones, E.; Fitzgerald, K. Pattern recognition receptors and the innate immune response to viral infection. Viruses 2011, 3, 920–940. [Google Scholar] [CrossRef] [Green Version]
- Chazal, M.; Beauclair, G.; Gracias, S.; Najburg, V.; Simon-Lorière, E.; Tangy, F.; Komarova, A.V.; Jouvenet, N. RIG-I recognizes the 5′ region of Dengue and Zika virus genomes. Cell Rep. 2018, 24, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Hirai, R.; Loo, Y.-M.; Owen, D.; Johnson, C.L.; Sinha, S.C.; Akira, S.; Fujita, T.; Gale, M. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc. Natl. Acad. Sci. USA 2007, 104, 582–587. [Google Scholar] [CrossRef] [Green Version]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730. [Google Scholar] [CrossRef]
- Wies, E.; Wang, M.K.; Maharaj, N.P.; Chen, K.; Zhou, S.; Finberg, R.W.; Gack, M.U. Dephosphorylation of the RNA sensors RIG-I and MDA5 by the phosphatase PP1 is essential for innate immune signaling. Immunity 2013, 38, 437–449. [Google Scholar] [CrossRef] [Green Version]
- Gack, M.U.; Shin, Y.C.; Joo, C.-H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916. [Google Scholar] [CrossRef]
- Okamoto, M.; Kouwaki, T.; Fukushima, Y.; Oshiumi, H. Regulation of RIG-I activation by K63-linked polyubiquitination. Front. Immunol. 2018, 8, 1942. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Dong, X.; He, Z.; Wu, Y.; Zhang, S.; Lin, J.; Yang, Y.; Chen, J.; An, S.; Yin, Y. Zika virus antagonizes interferon response in patients and disrupts RIG-I–MAVS interaction through its CARD-TM domains. Cell Biosci. 2019, 9, 46. [Google Scholar] [CrossRef]
- Wu, Y.; Liu, Q.; Zhou, J.; Xie, W.; Chen, C.; Wang, Z.; Yang, H.; Cui, J. Zika virus evades interferon-mediated antiviral response through the co-operation of multiple nonstructural proteins in vitro. Cell Discov. 2017, 3, 17006. [Google Scholar] [CrossRef]
- Ma, J.; Ketkar, H.; Geng, T.; Lo, E.; Wang, L.; Xi, J.; Sun, Q.; Zhu, Z.; Cui, Y.; Yang, L. Zika virus non-structural protein 4A blocks the RLR-MAVS signaling. Front. Microbiol. 2018, 9, 1350. [Google Scholar] [CrossRef] [Green Version]
- Jones, M.; Davidson, A.; Hibbert, L.; Gruenwald, P.; Schlaak, J.; Ball, S.; Foster, G.R.; Jacobs, M. Dengue virus inhibits alpha interferon signaling by reducing STAT2 expression. J. Virol. 2005, 79, 5414–5420. [Google Scholar] [CrossRef] [Green Version]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.-Y.; García-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef] [Green Version]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sánchez-Seco, M.P.; Evans, M.J.; Best, S.M. Zika virus targets human STAT2 to inhibit type I interferon signaling. Cell Host. Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [Green Version]
- Kumar, A.; Hou, S.; Airo, A.M.; Limonta, D.; Mancinelli, V.; Branton, W.; Power, C.; Hobman, T.C. Zika virus inhibits type-I interferon production and downstream signaling. EMBO Rep. 2016, 17, 1766–1775. [Google Scholar] [CrossRef] [Green Version]
- Fontaine, K.A.; Leon, K.E.; Khalid, M.M.; Tomar, S.; Jimenez-Morales, D.; Dunlap, M.; Kaye, J.A.; Shah, P.S.; Finkbeiner, S.; Krogan, N.J. The cellular NMD pathway restricts Zika virus infection and is targeted by the viral capsid protein. MBio 2018, 9, e02126-18. [Google Scholar] [CrossRef] [Green Version]
- Airo, A.M.; Urbanowski, M.D.; Lopez-Orozco, J.; You, J.H.; Skene-Arnold, T.D.; Holmes, C.; Yamshchikov, V.; Malik-Soni, N.; Frappier, L.; Hobman, T.C. Expression of flavivirus capsids enhance the cellular environment for viral replication by activating Akt-signalling pathways. Virology 2018, 516, 147–157. [Google Scholar] [CrossRef]
- Samuel, G.H.; Wiley, M.R.; Badawi, A.; Adelman, Z.N.; Myles, K.M. Yellow fever virus capsid protein is a potent suppressor of RNA silencing that binds double-stranded RNA. Proc. Natl. Acad. Sci. USA 2016, 113, 13863–13868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, R.; Singh, N.; Abdin, M.Z.; Patel, A.H.; Medigeshi, G.R. Dengue virus capsid interacts with DDX3X–a potential mechanism for suppression of antiviral functions in dengue infection. Front. Cell. Infect. Microbiol. 2018, 7, 542. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Wang, M.; Chen, S.; Cheng, A. The role of capsid in the flaviviral life cycle and perspectives for vaccine development. Vaccine 2020, 38, 6872–6881. [Google Scholar] [CrossRef] [PubMed]
- Colpitts, T.M.; Barthel, S.; Wang, P.; Fikrig, E. Dengue virus capsid protein binds core histones and inhibits nucleosome formation in human liver cells. PLoS ONE 2011, 6, e24365. [Google Scholar] [CrossRef] [Green Version]
- Urbanowski, M.D.; Hobman, T.C. The West Nile virus capsid protein blocks apoptosis through a phosphatidylinositol 3-kinase-dependent mechanism. J. Virol. 2013, 87, 872–881. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Turner, A.; Aggarwal, P.; Matter, A.; Storvick, E.; Arnett, D.K.; Broeckel, U. Comprehensive evaluation of AmpliSeq transcriptome, a novel targeted whole transcriptome RNA sequencing methodology for global gene expression analysis. BMC Genom. 2015, 16, 1069. [Google Scholar] [CrossRef] [Green Version]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [Green Version]
- Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008, 37, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Mi, H.; Muruganujan, A.; Ebert, D.; Huang, X.; Thomas, P.D. PANTHER version 14: More genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucleic Acids Res. 2018, 47, D419–D426. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25. [Google Scholar] [CrossRef] [Green Version]
- Consortium, G.O. The gene ontology resource: 20 years and still GOing strong. Nucleic Acids Res. 2018, (D1), D330–D338. [Google Scholar]
- Zou, W.; Zhang, D.-E. The interferon-inducible ubiquitin-protein isopeptide ligase (E3) EFP also functions as an ISG15 E3 ligase. J. Biol. Chem. 2006, 281, 3989–3994. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, S.K.; Dang, J.; Qin, Y.; Lichinchi, G.; Bansal, V.; Rana, T.M. Zika virus infection reprograms global transcription of host cells to allow sustained infection. Emerg. Microbes Infect. 2017, 6, e24. [Google Scholar] [CrossRef] [Green Version]
- Sun, X.; Hua, S.; Chen, H.-R.; Ouyang, Z.; Einkauf, K.; Tse, S.; Ard, K.; Ciaranello, A.; Yawetz, S.; Sax, P. Transcriptional changes during naturally acquired Zika virus infection render dendritic cells highly conducive to viral replication. Cell Rep. 2017, 21, 3471–3482. [Google Scholar] [CrossRef] [Green Version]
- Limonta, D.; Jovel, J.; Kumar, A.; Airo, A.; Hou, S.; Saito, L.; Branton, W.; Ka-Shu Wong, G.; Mason, A.; Power, C. Human Fetal Astrocytes Infected with Zika Virus Exhibit Delayed Apoptosis and Resistance to Interferon: Implications for Persistence. Viruses 2018, 10, 646. [Google Scholar] [CrossRef] [Green Version]
- Kozak, R.; Majer, A.; Biondi, M.; Medina, S.; Goneau, L.; Sajesh, B.; Slota, J.; Zubach, V.; Severini, A.; Safronetz, D. MicroRNA and mRNA dysregulation in astrocytes infected with Zika virus. Viruses 2017, 9, 297. [Google Scholar] [CrossRef] [Green Version]
- Breckpot, K.; Escors, D.; Arce, F.; Lopes, L.; Karwacz, K.; Van Lint, S.; Keyaerts, M.; Collins, M. HIV-1 lentiviral vector immunogenicity is mediated by Toll-like receptor 3 (TLR3) and TLR7. J. Virol. 2010, 84, 5627–5636. [Google Scholar] [CrossRef] [Green Version]
- Brown, B.D.; Sitia, G.; Annoni, A.; Hauben, E.; Sergi, L.S.; Zingale, A.; Roncarolo, M.G.; Guidotti, L.G.; Naldini, L. In vivo administration of lentiviral vectors triggers a type I interferon response that restricts hepatocyte gene transfer and promotes vector clearance. Blood 2006, 109, 2797–2805. [Google Scholar] [CrossRef] [Green Version]
- Yanai, H.; Chiba, S.; Hangai, S.; Kometani, K.; Inoue, A.; Kimura, Y.; Abe, T.; Kiyonari, H.; Nishio, J.; Taguchi-Atarashi, N. Revisiting the role of IRF3 in inflammation and immunity by conditional and specifically targeted gene ablation in mice. Proc. Natl. Acad. Sci. USA 2018, 115, 5253–5258. [Google Scholar] [CrossRef] [Green Version]
- Martín-Vicente, M.; Medrano, L.M.; Resino, S.; García-Sastre, A.; Martínez, I. TRIM25 in the Regulation of the Antiviral Innate Immunity. Front. Immunol. 2017, 8, 1187. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Aparicio, M.T.; Feinman, L.J.; García-Sastre, A.; Shaw, M.L. Paramyxovirus V proteins interact with the RIG-I/TRIM25 regulatory complex and inhibit RIG-I signaling. J. Virol. 2018, 92, e01960-17. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Heaton, S.M.; Parrish, N.F. Mammalian antiviral systems directed by small RNA. PLoS Pathog. 2021, 17, e1010091. [Google Scholar] [CrossRef]
- Soulat, D.; Bürckstümmer, T.; Westermayer, S.; Goncalves, A.; Bauch, A.; Stefanovic, A.; Hantschel, O.; Bennett, K.L.; Decker, T.; Superti-Furga, G. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 2008, 27, 2135–2146. [Google Scholar] [CrossRef] [Green Version]
- Schroder, M.; Baran, M.; Bowie, A.G. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008, 27, 2147–2157. [Google Scholar] [CrossRef]
- Mulhern, O.; Bowie, A.G. Unexpected roles for DEAD-box protein 3 in viral RNA sensing pathways. Eur. J. Immunol. 2010, 40, 933–935. [Google Scholar] [CrossRef]
- Oshiumi, H.; Sakai, K.; Matsumoto, M.; Seya, T. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. Eur. J. Immunol. 2010, 40, 940–948. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.-S.; Huang, I.-C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; García-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host. Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef] [Green Version]
- Faustino, A.F.; Martins, A.S.; Karguth, N.; Artilheiro, V.; Enguita, F.J.; Ricardo, J.C.; Santos, N.C.; Martins, I.C. Structural and Functional Properties of the Capsid Protein of Dengue and Related Flavivirus. Int. J. Mol. Sci. 2019, 20, 3870. [Google Scholar] [CrossRef] [Green Version]
- Sangiambut, S.; Keelapang, P.; Aaskov, J.; Puttikhunt, C.; Kasinrerk, W.; Malasit, P.; Sittisombut, N. Multiple regions in dengue virus capsid protein contribute to nuclear localization during virus infection. J. Gen. Virol. 2008, 89 Pt 5, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.; Hobman, T.C. The helicase activity of DDX56 is required for its role in assembly of infectious West Nile virus particles. Virology 2012, 433, 226–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez-Aparicio, M.T.; Ayllon, J.; Leo-Macias, A.; Wolff, T.; Garcia-Sastre, A. Subcellular Localizations of RIG-I, TRIM25, and MAVS Complexes. J Virol 2017, 91, e01155-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaturro, P.; Stukalov, A.; Haas, D.A.; Cortese, M.; Draganova, K.; Płaszczyca, A.; Bartenschlager, R.; Götz, M.; Pichlmair, A. An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature 2018, 561, 253. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, T.; Zahoor, M.A.; Heryani, N.; Workenhe, S.T.; Nazli, A.; Kaushic, C. TRIM26 Facilitates HSV-2 Infection by Downregulating Antiviral Responses through the IRF3 Pathway. Viruses 2021, 13, 70. [Google Scholar] [CrossRef]
- Wang, P.; Zhao, W.; Zhao, K.; Zhang, L.; Gao, C. TRIM26 negatively regulates interferon-β production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLoS Pathog. 2015, 11, e1004726. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | Description | Fold Change |
|---|---|---|
| CCL5 | C-C Motif Chemokine Ligand 5 | −1363.33 |
| MX1 | MX Dynamin Like GTPase 1 | −1055.14 |
| OASL | 2’-5’-Oligoadenylate Synthetase Like | −956.56 |
| IFIT2 | Interferon Induced Protein with Tetratricopeptide Repeats 2 | −718.98 |
| IFIT3 | Interferon Induced Protein with Tetratricopeptide Repeats 3 | −694.00 |
| IFIT1 | Interferon Induced Protein with Tetratricopeptide Repeats 1 | −363.25 |
| BST2 | Bone Marrow Stromal Cell Antigen 2 | −352.47 |
| ISG15 | ISG15 Ubiquitin-Like Modifier | −221.56 |
| PLEKHA4 | Pleckstrin Homology Domain Containing A4 | −212.54 |
| IFI6 | Interferon Alpha Inducible Protein 6 | −185.33 |
| HSPA6 | Heat Shock Protein Family A (Hsp70) Member 6 | −159.88 |
| IRF7 | Interferon Regulatory Factor 7 | −117.75 |
| IFIH1 | Interferon Induced with Helicase C Domain 1 | −111.11 |
| UBA7 | Ubiquitin Like Modifier Activating Enzyme 7 | −108.42 |
| HLA-B | Major Histocompatibility Complex, Class I, B | −95.93 |
| ATF3 | Activating Transcription Factor 3 | −88.65 |
| DDX58 | DExD/H-Box Helicase 58 | −79.13 |
| PARP10 | Poly (ADP-Ribose) Polymerase Family Member 10 | −73.80 |
| SAMD9L | Sterile Alpha Masotif Domain Containing 9 Like | −68.68 |
| SP110 | SP110 Nuclear Body Protein | −67.85 |
| Gene ID | Description | Fold Change |
|---|---|---|
| PRELID2 | PRELI Domain Containing 2 | 14.96 |
| ATP5MD | ATP Synthase Membrane Subunit DAPIT | 11.83 |
| SEC31A | SEC31 Homolog A, COPII Coat Complex Component | 10.48 |
| TXNDC5 | Thioredoxin Domain Containing 5 | 10.13 |
| ANKS4B | Ankyrin Repeat and Sterile Alpha Motif Domain Containing 4B | 8.24 |
| TBC1D22A | TBC1 Domain Family Member 22A | 5.22 |
| ITGB3BP | Integrin Subunit Beta 3 Binding Protein | 4.80 |
| SCARNA3 | Small Cajal Body-Specific RNA 3 | 4.20 |
| DDC | Dopa Decarboxylase | 4.20 |
| PIK3C2A | Phosphatidylinositol-4-Phosphate 3-Kinase Catalytic Subunit Type 2 Alpha | 3.99 |
| CENPE | Centromere Protein E | 3.96 |
| PLPPR1 | Phospholipid Phosphatase Related 1 | 3.80 |
| TMEM107 | Transmembrane Protein 107 | 3.70 |
| PMS2 | PMS1 Homolog 2, Mismatch Repair System Component | 3.63 |
| CDC25C | Cell Division Cycle 25C | 3.61 |
| EFCAB10 | EF-Hand Calcium Binding Domain 10 | 3.58 |
| NUP43 | Nucleoporin 43 | 3.25 |
| NONO | Non-POU Domain Containing Octamer Binding | 3.23 |
| PDK3 | Pyruvate Dehydrogenase Kinase 3 | 3.15 |
| HMMR | Hyaluronan Mediated Motility Receptor | 3.13 |
| Spectral | Counts | |||
|---|---|---|---|---|
| Protein ID | LacZ | Capsid (Exp 1) | Capsid (Exp 2) | Crapome Average |
| Q99733|NP1L4 | 2 | 39 | 37 | 3.1 |
| Q01105|SET | 0 | 26 | 24 | 5.4 |
| Q9BY44|EIF2A | 0 | 16 | 21 | 2.9 |
| 6P158|DHX57 | 1 | 15 | 15 | 2 |
| Q14258|TRIM25 | 0 | 16 | 13 | 1.9 |
| Q96GA3|LTV1 | 1 | 13 | 14 | 2.7 |
| Q9Y6M1|IF2B2 | 1 | 13 | 13 | 3.4 |
| Q8WU90|ZC3HF | 1 | 13 | 12 | 3.1 |
| O15355|PPM1G | 0 | 15 | 10 | 2 |
| Q12797|ASPH | 0 | 12 | 12 | 1.5 |
| Q9NX00|TM160 | 0 | 10 | 12 | 1 |
| Q99543|DNJC2 | 0 | 13 | 5 | 1.3 |
| Q13144|EI2BE | 0 | 9 | 9 | 1.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Airo, A.M.; Felix-Lopez, A.; Mancinelli, V.; Evseev, D.; Lopez-Orozco, J.; Shire, K.; Paszkowski, P.; Frappier, L.; Magor, K.E.; Hobman, T.C. Flavivirus Capsid Proteins Inhibit the Interferon Response. Viruses 2022, 14, 968. https://doi.org/10.3390/v14050968
Airo AM, Felix-Lopez A, Mancinelli V, Evseev D, Lopez-Orozco J, Shire K, Paszkowski P, Frappier L, Magor KE, Hobman TC. Flavivirus Capsid Proteins Inhibit the Interferon Response. Viruses. 2022; 14(5):968. https://doi.org/10.3390/v14050968
Chicago/Turabian StyleAiro, Adriana M., Alberto Felix-Lopez, Valeria Mancinelli, Danyel Evseev, Joaquin Lopez-Orozco, Kathy Shire, Patrick Paszkowski, Lori Frappier, Katharine E. Magor, and Tom C. Hobman. 2022. "Flavivirus Capsid Proteins Inhibit the Interferon Response" Viruses 14, no. 5: 968. https://doi.org/10.3390/v14050968
APA StyleAiro, A. M., Felix-Lopez, A., Mancinelli, V., Evseev, D., Lopez-Orozco, J., Shire, K., Paszkowski, P., Frappier, L., Magor, K. E., & Hobman, T. C. (2022). Flavivirus Capsid Proteins Inhibit the Interferon Response. Viruses, 14(5), 968. https://doi.org/10.3390/v14050968