
SARS-CoV-2 Lineage A.27: New Data from African Countries and Dynamics in the Context of the COVID-19 Pandemic

 ,
,  , ,
, ,  ,
for the REPAIR Consortium
,
for the REPAIR Consortium
Abstract
:1. Introduction
2. Materials and Methods
2.1. NGS Sequencing
2.2. Genome Assembly
2.3. Sequences Retrieved from GISAID Database
2.4. Phylogenetic Analysis
2.5. The Phylogeography Construction
3. Results
3.1. Most Recent Common Ancestor (tMRCA)
3.2. Phylogeography Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
- Marc Noguera i Julian, et al., Laboratori de Retrovirologia—irsiCaixa, HospitalUniversitariGermans Trias i Pujol, Crta. Canyet s/n, planta 2a, Edifici de la Maternitat08916 Badalona, Catalunya, Spain.
- Robert Dyrdak et al., Department of Clinical Microbiology, Karolinska University Hospital SE-171 76 Stockholm and Department of Microbiology, Tumor and Cell Biology (MTC), KarolinskaInstitutet|SE-171 65 Solna.
- MounerouSalou, PharmD, et al., Faculté des sciences de la santé/Université de Lomé, Président du Conseil de Coopération Université de Lomé, AMR focal point, Head of AMR referenceLab/MoHTogo, Supervisor of National Reference Center for HIV &STIs, Tel.: +22-89-032-6506/msalou@univ-lome.tg.
- Stefan Schmutz, et al., Institut für Medizinische Virologie, Universität Zürich, Winterthurerstrasse 190, CH-8057 Zurich
- Nadeau Sarah Ann et al., Department of Biosystems Science and Engineering, ETH Zürich, Basel, Switzerland Swiss and Institute of Bioinformatics, Switzerland
- MišaKorva et al., Univerza v Ljubljani|Medicinskafakulteta INŠTITUT ZA Mikrobiologijo In Imunologijo, University of Ljubljana | Faculty of Medicine, Institute of Microbiology and Immunology, Zaloška 4, SI—1000 Ljubljana, Slovenia.
- Bas Oude Munnink Erasmus MC, et al., Department of Viroscience, WHO Collaborating Centre for Arbovirus and Viral Hemorrhagic Fever Reference and Research, Rotterdam, the Netherlands.
- YLeonMutesa et al., Center for Human Genetics, College of Medicine and Health Sciences, University of Rwanda, Kigali, Rwanda.
- Keith Durkin et al., Laboratory of Human Genetics, GIGA Research Institute, Liège, Belgium.
- Guy Baele et al., Department of Microbiology, Immunology and Transplantation, Rega Institute, KU Leuven, Leuven, Belgium.
- Nadine Rujeni et al., School of Health Sciences, College of Medicine and Health Sciences, University of Rwanda, Kigali, Rwanda
- YvanButera et al., Center for Human Genetics, College of Medicine and Health Sciences, University of Rwanda, Kigali, Rwanda; Laboratory of Human Genetics, GIGA Research Institute, Liège, Belgium
- Gabriel Gonzalez et al., UCD National Virus Reference Laboratory, Dublin, Ireland
- Angela Di Martino et al., DipartimentoMalattieInfettive, IstitutoSuperiore di Sanità, Viale Regina Elena, 299, 00161 Roma, Italia, Tel.: +(39) 0649903332/2667
- Piet Maes et al., Laboratory of Clinical and Epidemiological Virology (Rega Institute), Rega—Herestraat 49—box 1040, 3000 Leuven
- Emmanouil I. Athanasiadis, et al., Computational Biology Unit (CBU)|Greek Genome Center, Biomedical Research Foundation|Academy Of Athens, 4 SoranouEphessiou Street|115 27|Athens|Greece mathan@bioacademy.gr, http://www.bioacademy.gr/ (accessed on 12 November 2021).
- José MaríaMarimón et al., Microbiology Department, University Hospital Donostia- Biodonostia Health Research Institute, Paseo del Beguristain s/n, 20014 Donostia-San Sebastián, Spain Tel.: +34-943007046 (ext. 833189) Email: josemaria.marimonortizdez@osakidetza.eus
- Serge Alain SADEUH-MBA et al.,Virology Service, Centre Pasteur du Cameroun 451, Rue 2005, Yaounde 2, PO box 1274, Yaounde, Cameroon Tel: +237-222-231-803, Mobile: +237-677-966-664 Email: sadeuh@pasteur-yaounde.org/http://www.pasteur-yaounde.org (accessed on 12 November 2021).
- SlimFourati et al., PU-PH, Laboratoire de Virologie, Centre National de Référence des Hépatites B, C et delta, INSERM U955, Hôpital Henri Mondor, 51 avenue du Maréchal de Lattre de Tassigny 94010 Créteil, France, Tel.: +(33)-14-517-8145, Fax: +(33)-14-981-2839.
- Bruno Lina et al., Laboratoire de virologie, Institut des Agents Infectieux, CHU de Lyon HCL—GH Nord-Hôpital de la Croix Rousse, CNR Virus des Infections Respiratoires—France SUD, 103 Grande Rue de la Croix Rousse, 69317 Lyon CEDEX 4, France.
- Jonas Fuchs et al., Institute of Virology, Freiburg University Medical Center, Faculty of Medicine, University of Freiburg, Germany.
- Sylvie Van Der Werf and Vincent Enouf et al., National Reference Center for Viruses of Respiratory infections, Institut Pasteur de Paris.
Conflicts of Interest
References
- Wise, J. COVID-19: New coronavirus variant is identified in UK. BMJ 2020, 371, m4857. [Google Scholar] [CrossRef] [PubMed]
- The World Health Organisation (WHO). Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/en/activities/tracking-SARS-CoV-2-variants/ (accessed on 12 November 2021).
- Tegally, H.; Wilkinson, E.; Althaus, C.L.; Giovanetti, M.; San, J.E.; Giandhari, J.; Pillay, S.; Naidoo, Y.; Ramphal, U.; Msomi, N.; et al. Rapid replacement of the Beta variant by the Delta variant in South Africa. medRxiv 2021. [Google Scholar] [CrossRef]
- Earnest, R.; Uddin, R.; Matluk, N.; Renzette, N.; Siddle, K.J.; Loreth, C.; Adams, G.; Tomkins-Tinch, C.H.; Petrone, M.E.; Rothman, J.E.; et al. Comparative transmissibility of SARS-CoV-2 variants Delta and Alpha in New England, USA. medRxiv 2021. [Google Scholar] [CrossRef]
- Lineage List. Available online: https://cov-lineages.org/lineage_list.html (accessed on 12 November 2021).
- SARS-CoV-2 (hCoV-19) Mutation Reports. Available online: https://outbreak.info/situation-reports (accessed on 12 November 2021).
- European Centre for Disease Prevention and Control. SARS-CoV-2 Variants of Concern. Available online: https://www.ecdc.europa.eu/en/covid-19/variants-concern (accessed on 12 November 2021).
- COV-UK. Investigation of SARS-CoV-2 Variants of Concern: Technical Briefings. Available online: https://www.gov.uk/government/publications/investigation-of-novel-sars-cov-2-variant-variant-of-concern-20201201 (accessed on 12 November 2021).
- Available online: https://www.sfm-microbiologie.org/wp-content/uploads/2021/04/Tableau-Variants-SARS-CoV-2-_SFM-AC43-V3b_010421.pdf (accessed on 12 November 2021).
- Calvignac-Spencer, S.; Budt, M.; Huska, M.; Richard, H.; Leipold, L.; Grabenhenrich, L.; Semmler, T.; Von Kleist, M.; Kröger, S.; Wolff, T.; et al. Rise and Fall of SARS-CoV-2 Lineage A.27 in Germany. Viruses 2021, 13, 1491. [Google Scholar] [CrossRef] [PubMed]
- Fourati, S.; Decousser, J.W.; Khouider, S.; N’Debi, M.; Demontant, V.; Trawinski, E.; Gourgeon, A.; Gangloff, C.; Destras, G.; Bal, A.; et al. Novel SARS-CoV-2 Variant Derived from Clade 19B, France. Emerg. Infect. Dis. 2021, 5, 1540–1543. [Google Scholar] [CrossRef] [PubMed]
- Anoh, E.A.; Schubert, G.; Wayoro, O.; Pacôme, M.; Belarbi, E.; Sachse, A.; Calvignac-Spencer, S.; Leendertz, F.; Diané, B.; Akoua-Koffi, C. SARS-CoV-2 variants of concern, variants of interest and lineage A.27 are on the rise in Côte d’Ivoire. medRxiv 2021. [Google Scholar] [CrossRef]
- Colson, P.; Levasseur, A.; Delerce, J.; Pinault, L.; Dudouet, P.; Devaux, C.; Fournier, P.E.; La Scola, B.; Lagier, J.C.; Raoult, D. Spreading of a new SARS-CoV-2 N501Y spike variant in a new lineage. ClinMicrobiol Infect. 2021, 27, 1352. [Google Scholar] [CrossRef]
- Greaney, A.J.; Starr, T.N.; Barnes, C.O.; Weisblum, Y.; Schmidt, F.; Caskey, M.; Gaebler, C.; Cho, A.; Agudelo, M.; Finkin, S.; et al. Mapping mutations to the SARS-CoV-2 RBD that escape binding by different classes of antibodies. Nat. Commun. 2021, 12, 4196. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Plante, K.S.; Plante, J.A.; Xie, X.; Zhang, X.; Ku, Z.; An, Z.; Scharton, D.; Schindewolf, C.; et al. The N501Y spike substitution enhances SARS-CoV-2 infection and transmission. Nature 2022, 602, 294–299. [Google Scholar] [CrossRef]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, infectivity, and antibody neutralization of an emerging SARS-CoV-2 variant in California carrying a L452R spike protein mutation. medRxiv 2021. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef] [PubMed]
- GISAID Initiative. Available online: https://www.gisaid.org/ (accessed on 12 November 2021).
- Bhoyar, R.C.; Jain, A.; Sehgal, P.; Divakar, M.K.; Sharma, D.; Imran, M.; Jolly, B.; Ranjan, G.; Rophina, M.; Sharma, S.; et al. High throughput detection and genetic epidemiology of SARS-CoV-2 using COVID Seq next-generation sequencing. PLoS ONE 2021, 16, e0247115. [Google Scholar] [CrossRef] [PubMed]
- Danecek, P.; Bonfield, J.K.; Liddle, J.; Marshall, J.; Ohan, V.; Pollard, M.O.; Whitwham, A.; Keane, T.; McCarthy, S.A.; Davies, R.M.; et al. Twelve years of SAMtools and BCFtools. GigaScience 2021, 10, giab008. [Google Scholar] [CrossRef] [PubMed]
- EDGE Bioinformatics. Available online: https://edgebioinformatics.org/ (accessed on 12 November 2021).
- Lo, C.C.; Chain, P.S. Rapid evaluation and quality control of next generation sequencing data with FaQCs. BMC Bioinform. 2014, 15, 366. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997v2. [Google Scholar]
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. MolBiolEvol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Elbe, S.; Buckland-Merrett, G. Data, disease and diplomacy: GISAID’s innovative contribution to global health. Glob. Chall. 2017, 1, 33–46. [Google Scholar] [CrossRef] [Green Version]
- Alpert, T.; Brito, A.F.; Lasek-Nesselquist, E.; Rothman, J.; Valesano, A.L.; MacKay, M.J.; Petrone, M.E.; Breban, M.I.; Watkins, A.E.; Vogels, C.B.F.; et al. Early introductions and community transmission of SARS-CoV-2 variant B.1.1.7 in the United States. medRxiv 2021. [Google Scholar] [CrossRef]
- Simon-Loriere, E. Potential New Lineage Causing a Cluster in Mayotte 2021. Available online: https://github.com/cov-lineages/pango-designation/issues/11 (accessed on 12 November 2021).
- Kaleta, T.; Kern, L.; Hong, S.; Hölzer, M.; Kochs, G.; Beer, J.; Schnepf, D.; Schwemmle, M.; Bollen, N.; Kolb, P.; et al. Antibody escape and global spread of SARS-CoV-2 lineage A.27. Nat. Commun. 2022, 13, 1152. [Google Scholar] [CrossRef]
- Fares, W.; Ghedira, K.; Gdoura, M.; Chouikha, A.; Haddad-Boubaker, S.; Khedhiri, M.; Ayouni, K.; Lamari, A.; Touzi, H.; Hammemi, W.; et al. Sequencing Using a Two-Step Strategy Reveals High Genetic Diversity in the S Gene of SARS-CoV-2 after a High-Transmission Period in Tunis, Tunisia. Microbiol. Spectr. 2022, 9, e0063921. [Google Scholar] [CrossRef] [PubMed]
- Motozono, C.; Toyoda, M.; Zahradnik, J.; Saito, A.; Nasser, H.; Tan, T.S.; Ngare, I.; Kimura, I.; Uriu, K.; Kosugi, Y.; et al. Genotype to Phenotype Japan (G2P-Japan) Consortium, Ikeda T, Nakagawa S, Ueno T, Sato, K. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe 2021, 29, 1124–1136. [Google Scholar] [CrossRef] [PubMed]
- Papa, G.; Mallery, D.L.; Albecka, A.; Welch, L.G.; Cattin-Ortolá, J.; Luptak, J.; Paul, D.; McMahon, H.T.; Goodfellow, I.G.; Carter, A.; et al. Furin cleavage of SARS-CoV-2 Spike promotes but is not essential for infection and cell-cell fusion. PLoS Pathog. 2021, 17, e1009246. [Google Scholar] [CrossRef] [PubMed]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam Van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef] [PubMed]
- Hodcroft, E.B.; Domman, D.B.; Snyder, D.J.; Oguntuyo, K.Y.; Van Diest, M.; Densmore, K.H.; Schwalm, K.C.; Jennifer, L.J.F.; Rona, S.C.; Martha, M.S.; et al. Emergence in late 2020 of multiple lineages of SARS-CoV-2 Spike protein variants affecting amino acid position 677. medRxiv 2021. [Google Scholar] [CrossRef]
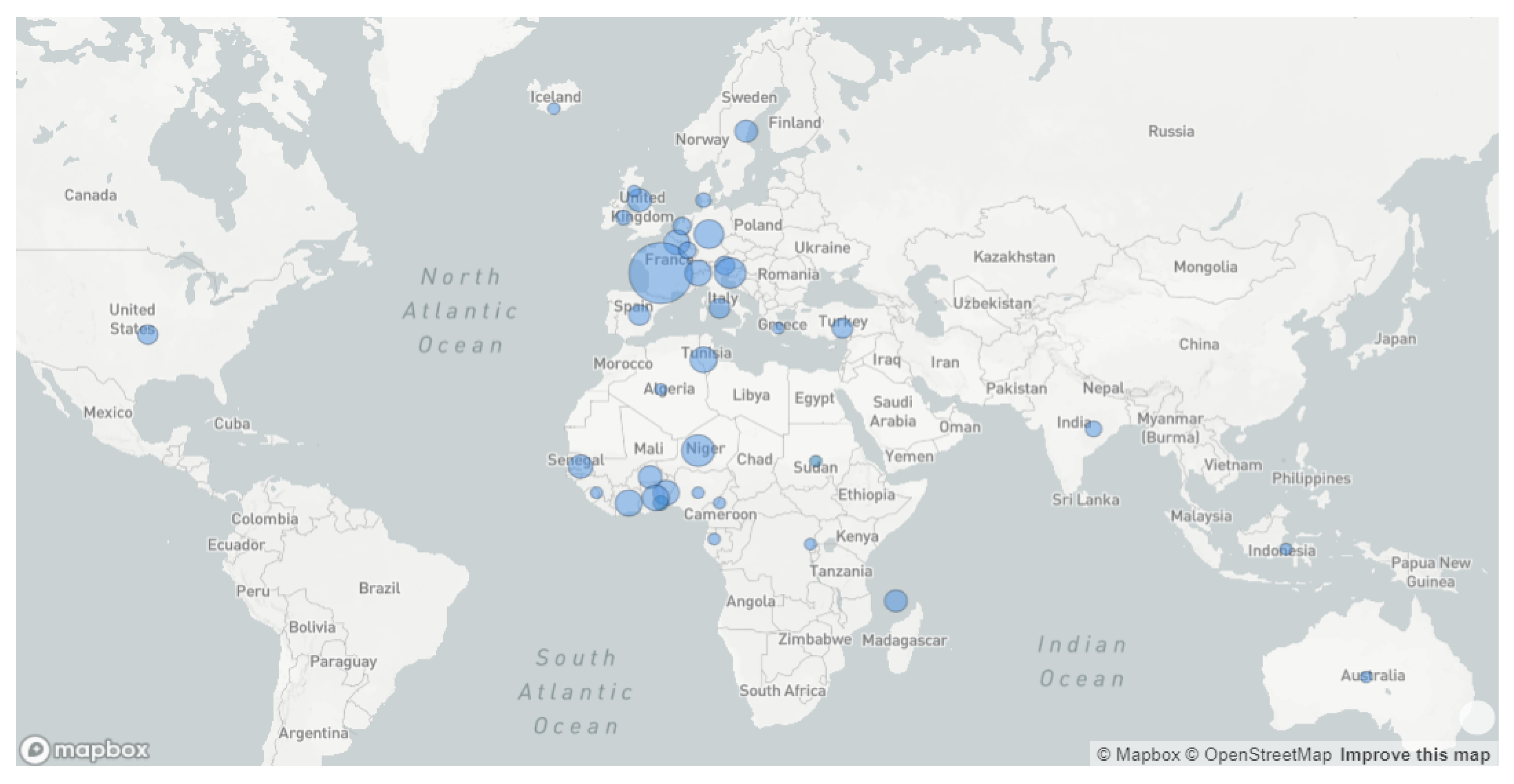
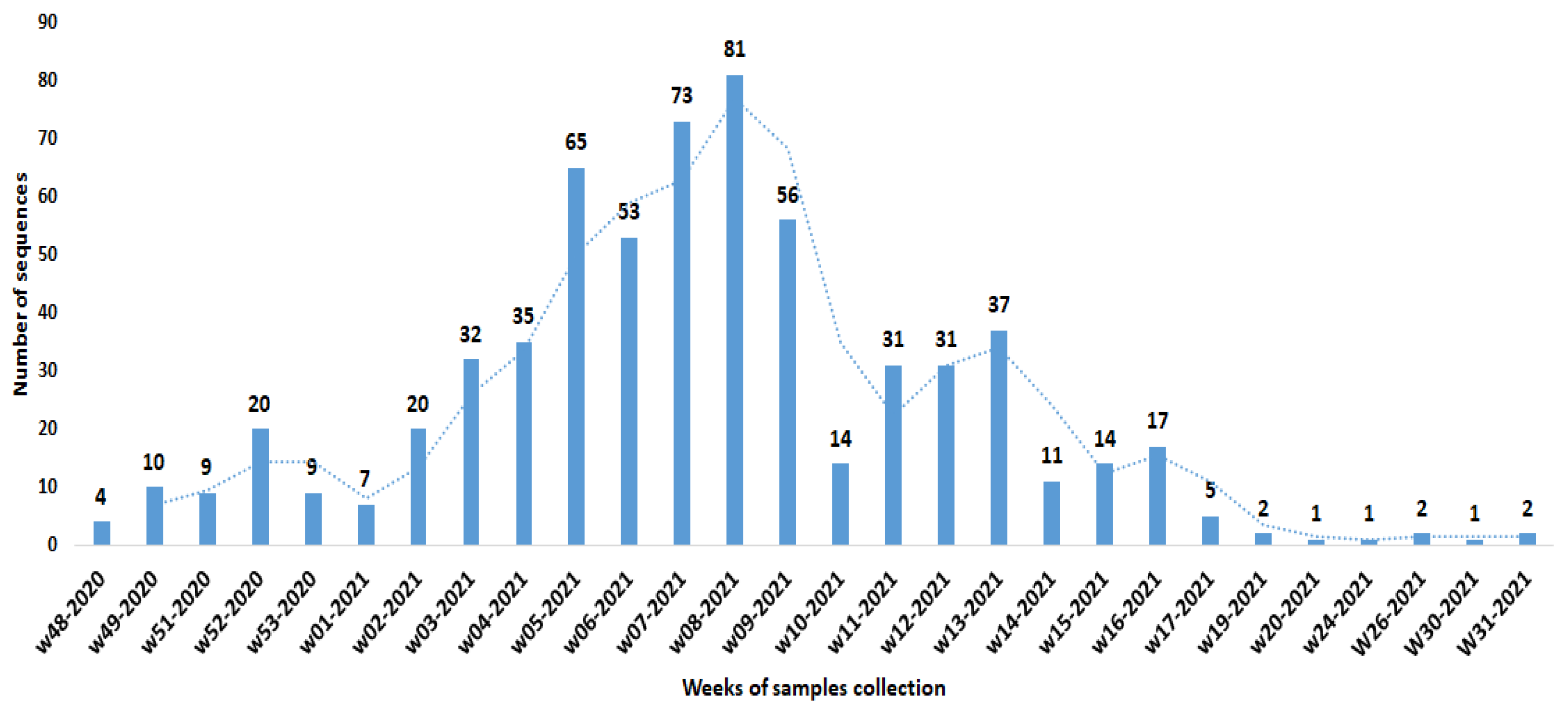
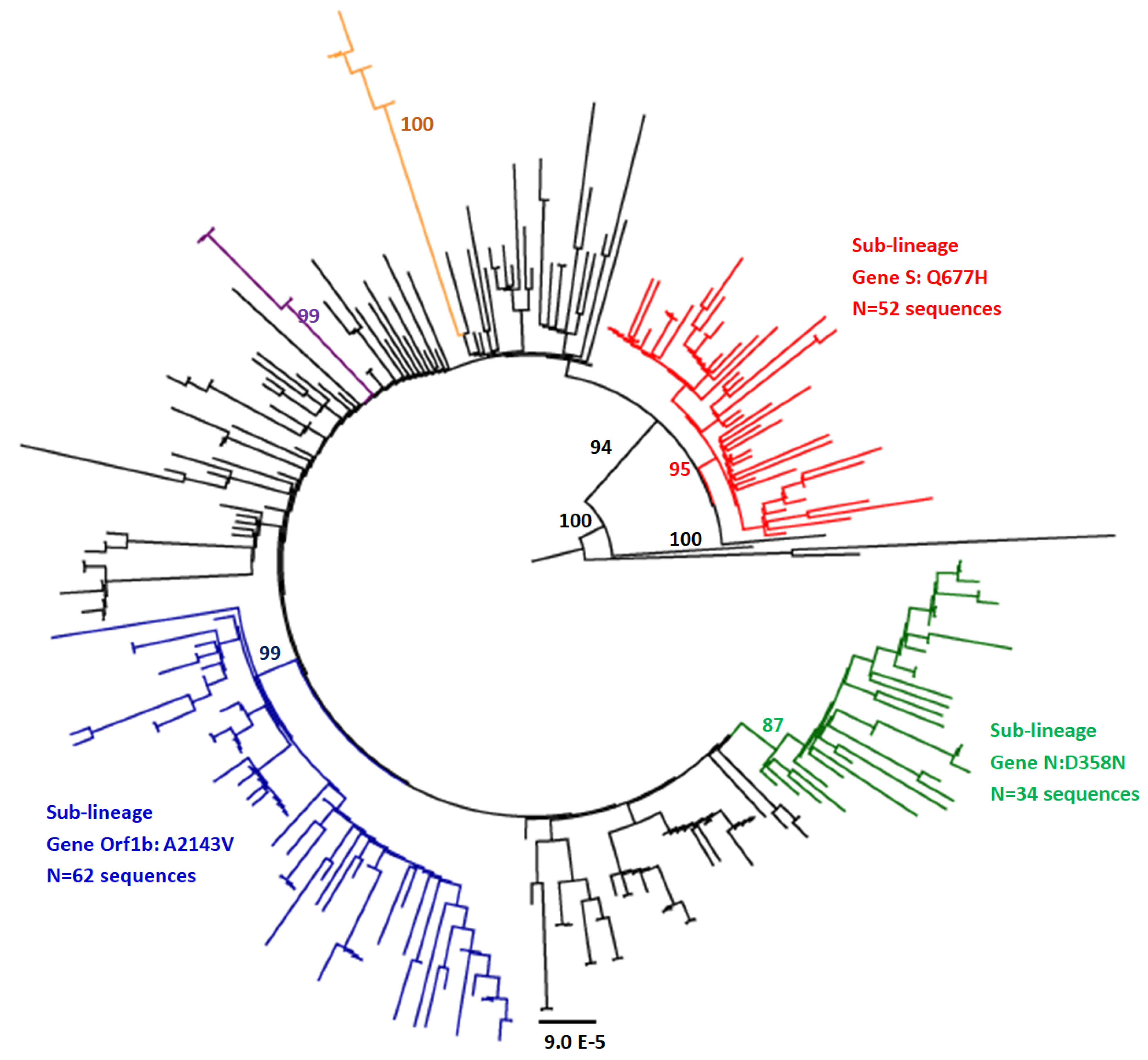


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Code | Country | Date of Isolation YYYY-MM-DD | ClinicalForm | Epidemiologicallink | Age (Years) | Gender | GISAID (Accession N°) |
|---|---|---|---|---|---|---|---|
| hCoV-19/Africa/Algeria/G46955 | Algeria | 2021-06-14 | Unknown | Indigenous | 66 | M | EPI_ISL_3946135 |
| hCoV-19/Africa/Cameroon/ CPC_21V-8285 | Cameroon | 2021-02-22 | Asymptomatic | Indigenous | 26 | F | EPI_ISL_8298662 |
| hCoV-19/Africa/Niger/EP40/20 | Niger | 2020-11-27 | Unknown | Indigenous | 27 | M | EPI_ISL_2281778 |
| hCoV-19/Africa/Niger/Niamey/EP67 | Niger | 2020-11-27 | Unknown | Indigenous | 25 | M | EPI_ISL_3804025 |
| hCoV-19/Africa/Niger/Niamey/EP75 | Niger | 2020-11-27 | Unknown | Indigenous | 28 | M | EPI_ISL_3804026 |
| hCoV-19/Africa/Niger/Niamey/CE5523 | Niger | 2020-11-28 | Unknown | Indigenous | 19 | M | EPI_ISL_2281839 |
| hCoV-19/Africa/Niger/EP132 | Niger | 2020-11-30 | Unknown | Indigenous | 29 | F | EPI_ISL_2281837 |
| hCoV-19/Africa/Niger/Niamey/CE1574 | Niger | 2020-11-30 | Unknown | Indigenous | 51 | M | EPI_ISL_2281838 |
| hCoV-19/Africa/Niger/Niamey/CE5715 | Niger | 2020-11-30 | Unknown | Indigenous | 30 | M | EPI_ISL_2281827 |
| hCoV-19/Africa/Niger/Niamey/17786 | Niger | 2020-11-31 | Asymtomatic | Indigenous | 40 | M | EPI_ISL_3804022 |
| hCoV-19/Africa/Niger/Agadez/Y/20 | Niger | 2020-12-04 | Unknown | Indigenous | M | EPI_ISL_2281821 | |
| hCoV-19/Africa/Niger/EP166 | Niger | 2020-12-04 | Unknown | Indigenous | 40 | M | EPI_ISL_2281787 |
| hCoV-19/Africa/Niger/EP185 | Niger | 2020-12-04 | Unknown | Indigenous | 32 | M | EPI_ISL_2281806 |
| hCoV-19/Africa/Niger/Niamey/EP117 | Niger | 2020-12-04 | Unknown | Indigenous | 38 | M | EPI_ISL_3804030 |
| hCoV-19/Africa/Niger/Niamey/12719 | Niger | 2020-12-06 | Asymptomatic | Indigenous | 60 | F | EPI_ISL_2281803 |
| hCoV-19/Africa/Niger/Niamey/12734 | Niger | 2020-12-06 | Asymptomatic | Indigenous | 37 | F | EPI_ISL_2281799 |
| hCoV-19/Africa/Niger/Agadez/76/20 | Niger | 2020-12-16 | Unknown | Indigenous | 34 | M | ** |
| hCoV-19/Africa/Niger/Niamey/8612 | Niger | 2020-12-17 | Unknown | Indigenous | <1 | M | EPI_ISL_3804031 |
| hCoV-19/Africa/Niger/Doutchi/8617 | Niger | 2020-12-22 | Unknown | Indigenous | 4 | M | EPI_ISL_2281791 |
| hCoV-19/Africa/Niger/Niamey/8637 | Niger | 2020-12-22 | Unknown | Indigenous | 8 | M | EPI_ISL_3804064 |
| hCoV-19/Africa/Niger/Niamey/16565 | Niger | 2020-12-24 | Asymptomatic | Indigenous | 66 | F | EPI_ISL_2281798 |
| hCoV-19/Africa/Niger/Niamey/8639 | Niger | 2020-12-24 | Unknown | Indigenous | <1 | F | EPI_ISL_3804036 |
| hCoV-19/Africa/Niger/Niamey/16781 | Niger | 2020-12-25 | Unknown | Indigenous | 35 | M | EPI_ISL_2281834 |
| hCoV-19/Africa/Niger/Agadez/18/20 | Niger | 2020-12-26 | Asymptomatic | Indigenous | 23 | M | EPI_ISL_2281813 |
| hCoV-19/Africa/Niger/Niamey/16924 | Niger | 2020-12-26 | Asymptomatic | Indigenous | 33 | F | ** |
| hCoV-19/Africa/Niger/Niamey/16930 | Niger | 2020-12-26 | Asymptomatic | Indigenous | 39 | M | EPI_ISL_3804057 |
| hCoV-19/Africa/Niger/Niamey/16933 | Niger | 2020-12-26 | Asymptomatic | Indigenous | 26 | F | EPI_ISL_3804032 |
| hCoV-19/Africa/Niger/Niamey/17065 | Niger | 2020-12-26 | Asymptomatic | Indigenous | 10 | F | EPI_ISL_3804033 |
| hCoV-19/Africa/Niger/Niamey/17120 | Niger | 2020-12-26 | Asymptomatic | Indigenous | 40 | M | EPI_ISL_3804022 |
| hCoV-19/Africa/Niger/Niamey/17198 | Niger | 2020-12-26 | Asymptomatic | Indigenous | 71 | M | EPI_ISL_3804019 |
| hCoV-19/Africa/Niger/Niamey/17204 | Niger | 2020-12-26 | Asymptomatic | Indigenous | 4 | M | EPI_ISL_3804020 |
| hCoV-19/Africa/Niger/Niamey/17208 | Niger | 2020-12-26 | Asymptomatic | Indigenous | 62 | M | EPI_ISL_3804021 |
| hCoV-19/Africa/Niger/Niamey/8662 | Niger | 2020-12-30 | Unknown | Indigenous | <1 | M | EPI_ISL_2281815 |
| hCoV-19/Africa/Niger/Gaya/8668 | Niger | 2020-12-31 | Unknown | Indigenous | 13 | M | EPI_ISL_2281777 |
| hCoV-19/Africa/Niger/Niamey/17751 | Niger | 2020-12-31 | Asymtomatic | Indigenous | 35 | M | EPI_ISL_2281789 |
| hCoV-19/Africa/Niger/Niamey/17753 | Niger | 2020-12-31 | Asymtomatic | Indigenous | 27 | M | EPI_ISL_2281775 |
| hCoV-19/Africa/Niger/Niamey/17755 | Niger | 2020-12-31 | Asymtomatic | Indigenous | 56 | M | EPI_ISL_3804058 |
| hCoV-19/Africa/Niger/Niamey/17827 | Niger | 2021-01-01 | Asymtomatic | Indigenous | 66 | M | EPI_ISL_3804059 |
| hCoV-19/Africa/Niger/Niamey/CE1770 | Niger | 2021-01-09 | Asymtomatic | Indigenous | 29 | M | EPI_ISL_2281782 |
| hCoV-19/Africa/Niger/Niamey/CNU57 | Niger | 2021-01-09 | Unknown | Indigenous | 40 | F | EPI_ISL_3804035 |
| hCoV-19/Africa/Niger/CNU180 | Niger | 2021-01-19 | Unknown | Indigenous | 47 | M | EPI_ISL_3804063 |
| hCoV-19/Africa/Niger/Niamey/19027 | Niger | 2021-01-19 | Mild | Indigenous | 39 | M | EPI_ISL_3804060 |
| hCoV-19/Africa/Niger/Niamey/19030 | Niger | 2021-01-19 | Mild | Indigenous | 47 | M | EPI_ISL_3804061 |
| hCoV-19/Africa/Niger/Niamey/19031 | Niger | 2021-01-19 | Mild | Indigenous | 64 | M | EPI_ISL_3804062 |
| hCoV-19/Africa/Niger/Niamey/CE4805 | Niger | 2021-01-22 | Unknown | Indigenous | 42 | F | EPI_ISL_3804068 |
| hCoV-19/Africa/Niger/Niamey/CE2069 | Niger | 2021-02-01 | Unknown | Indigenous | M | EPI_ISL_3804066 | |
| hCoV-19/Africa/Niger/Niamey/CE729 | Niger | 2021-02-05 | Unknown | Indigenous | 63 | M | EPI_ISL_2281760 |
| hCoV-19/Africa/Niger/Niamey/19902 | Niger | 2021-02-17 | Asymptomatic | Indigenous | 69 | M | EPI_ISL_2281759 |
| hCoV-19/Africa/Senegal/156123/ | Senegal | 2021-01-22 | Asymptomatic | Unknown | 74 | M | xx |
| hCoV-19/Africa/Senegal/180227 | Senegal | 2021-02-17 | Asymptomatic | Unknown | 53 | M | xx |
| hCoV-19/Africa/Senegal/6941172_S6 | Senegal | 2021-04-12 | Asymptomatic | Indigenous | 31 | F | EPI_ISL_8528025 |
| hCoV-19/Africa/Senegal/6942249_S17 | Senegal | 2021-04-13 | Asymptomatic | Indigenous | 42 | M | EPI_ISL_8528036 |
| hCoV-19/Africa/Senegal/6944287_S22 | Senegal | 2021-04-15 | Asymptomatic | Indigenous | 30 | F | EPI_ISL_8528057 |
| hCoV-19/Africa/Senegal/6980248_S62 | Senegal | 2021-04-19 | Asymptomatic | Indigenous | 50 | F | EPI_ISL_8528097 |
| hCoV-19/Africa/Senegal/6981610_S73 | Senegal | 2021-04-20 | Asymptomatic | Indigenous | 55 | M | EPI_ISL_8528113 |
| hCoV-19/Africa/Senegal/6981616_S74 | Senegal | 2021-04-20 | Asymptomatic | Indigenous | 51 | M | EPI_ISL_8528114 |
| hCoV-19/Africa/Senegal/6986192_S79 | Senegal | 2021-04-21 | Asymptomatic | Indigenous | 34 | M | EPI_ISL_8528124 |
| hCoV-19/Africa/Senegal/6986443_S83 | Senegal | 2021-04-22 | Asymptomatic | Indigenous | 42 | M | EPI_ISL_8528128 |
| hCoV-19/Africa/Senegal/6986447_S84 | Senegal | 2021-04-22 | Asymptomatic | Indigenous | 52 | M | EPI_ISL_8528129 |
| hCoV-19/Africa/Senegal/6990626_S101 | Senegal | 2021-04-25 | Asymptomatic | Indigenous | 56 | M | EPI_ISL_8528160 |
| hCoV-19/Africa/Senegal/6990754_S100 | Senegal | 2021-04-25 | Asymptomatic | Chinese | 36 | M | EPI_ISL_8528161 |
| hCoV-19/Africa/Senegal/7914449_S260 | Senegal | 2021-05-20 | Asymptomatic | Chinese | 30 | F | EPI_ISL_8528306 |
| hCoV-19/Africa/Tunisia/Tunis/Q-5509 | Tunisia | 2021-02-02 | Mild | Indigenous | 27 | F | EPI_ISL_1208398 |
| hCoV-19/Africa/Tunisia/Tunis/Q-5516 | Tunisia | 2021-02-02 | Mild | Indigenous | 34 | F | EPI_ISL_12316665 |
| hCoV-19/Africa/Tunisia/Tunis/Q-6575 | Tunisia | 2021-02-07 | Mild | Indigenous | M | EPI_ISL_1207287 | |
| hCoV-19/Africa/Tunisia/Tunis/Q-6590 | Tunisia | 2021-02-07 | Mild | Indigenous | 17 | M | EPI_ISL_2035942 |
| hCoV-19/Africa/Tunisia/Tunis/Q-9052 | Tunisia | 2021-02-23 | Mild | Indigenous | 78 | M | EPI_ISL_12316666 |
| hCoV-19/Africa/Tunisia/Tunis/Q-9058 | Tunisia | 2021-02-23 | Mild | Indigenous | 79 | M | ** |
| hCoV-19/Africa/Tunisia/Tunis/Q-9081 | Tunisia | 2021-02-23 | Mild | Indigenous | 31 | M | EPI_ISL_12316667 |
| hCoV-19/Africa/Tunisia/Tunis/Q-9082 | Tunisia | 2021-02-23 | Mild | Indigenous | 85 | F | EPI_ISL_12316668 |
| hCoV-19/Africa/Tunisia/Tunis/U-4071 | Tunisia | 2021-02-24 | Asymptomatic | Cote d’Ivoire | 36 | M | xx |
| hCoV-19/Africa/Tunisia/Tunis/Q-9407 | Tunisia | 2021-02-25 | Mild | Indigenous | 32 | M | EPI_ISL_10101339 |
| hCoV-19/Africa/Tunisia/Tunis/Q-9581 | Tunisia | 2021-02-25 | Mild | Indigenous | 80 | M | EPI_ISL_12316669 |
| hCoV-19/Africa/Tunisia/Ariana/Q-9623 | Tunisia | 2021-02-25 | Mild | Indigenous | 25 | F | EPI_ISL_10101331 |
| hCoV-19/Africa/Tunisia/Siliana/Q-9993 | Tunisia | 2021-03-01 | Severe | Indigenous | 33 | M | EPI_ISL_12316670 |
| hCoV-19/Africa/Tunisia/Siliana/B-0386 | Tunisia | 2021-03-03 | Mild | Indigenous | 65 | F | xx |
| hCoV-19/Africa/Tunisia/Tunis/B-0396 | Tunisia | 2021-03-03 | Mild | Indigenous | 67 | F | EPI_ISL_2035946 |
| hCoV-19/Africa/Tunisia/Tunis/B-1388 | Tunisia | 2021-03-09 | Mild | Indigenous | 30 | F | xx |
| hCoV-19/Africa/Tunisia/Manouba/S-0159 | Tunisia | 2021-03-16 | Mild | Indigenous | 34 | F | EPI_ISL_10101347 |
| hCoV-19/Africa/Tunisia/Mednine/S-0418 | Tunisia | 2021-04-20 | Mild | Libya | 54 | M | xx |
| hCoV-19/Africa/Tunisia/Ben Arous/C-4468 | Tunisia | 2021-04-21 | Mild | Indigenous | 20 | F | EPI_ISL_10141399 |
| hCoV-19/Africa/Tunisia/Ben Arous/C-4469 | Tunisia | 2021-04-21 | Mild | Indigenous | 14 | M | EPI_ISL_10141512 |
| hCoV-19/Africa/Tunisia/Tunis/C-5524 | Tunisia | 2021-04-23 | Severe | Indigenous | 72 | M | EPI_ISL_10141507 |
| hCoV-19/Africa/Tunisia/Manouba/S-0042 | Tunisia | 2021-03-16 | Mild | Indigenous | 68 | F | EPI_ISL_12280586 |
| hCoV-19/Africa/Tunisia/Manouba/S-0043 | Tunisia | 2021-03-16 | Mild | Indigenous | 44 | M | EPI_ISL_10101222 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chouikha, A.; Lagare, A.; Ghedira, K.; Diallo, A.; Njouom, R.; Sankhe, S.; Derrar, F.; Victoir, K.; Dellagi, K.; Triki, H.; et al. SARS-CoV-2 Lineage A.27: New Data from African Countries and Dynamics in the Context of the COVID-19 Pandemic. Viruses 2022, 14, 1007. https://doi.org/10.3390/v14051007
Chouikha A, Lagare A, Ghedira K, Diallo A, Njouom R, Sankhe S, Derrar F, Victoir K, Dellagi K, Triki H, et al. SARS-CoV-2 Lineage A.27: New Data from African Countries and Dynamics in the Context of the COVID-19 Pandemic. Viruses. 2022; 14(5):1007. https://doi.org/10.3390/v14051007
Chicago/Turabian StyleChouikha, Anissa, Adamou Lagare, Kais Ghedira, Amadou Diallo, Richard Njouom, Safietou Sankhe, Fawzi Derrar, Kathleen Victoir, Koussay Dellagi, Henda Triki, and et al. 2022. "SARS-CoV-2 Lineage A.27: New Data from African Countries and Dynamics in the Context of the COVID-19 Pandemic" Viruses 14, no. 5: 1007. https://doi.org/10.3390/v14051007
APA StyleChouikha, A., Lagare, A., Ghedira, K., Diallo, A., Njouom, R., Sankhe, S., Derrar, F., Victoir, K., Dellagi, K., Triki, H., & Diagne, M. M., for the REPAIR Consortium. (2022). SARS-CoV-2 Lineage A.27: New Data from African Countries and Dynamics in the Context of the COVID-19 Pandemic. Viruses, 14(5), 1007. https://doi.org/10.3390/v14051007