
HDAC Inhibitors Enhance Efficacy of the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T in Pancreatic and Triple-Negative Breast Cancer Models


 , ,
, , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Viruses and Inhibitors
2.2. Cell Viability Assay and Synergistic Interactions
2.3. Viral Replication Assay by Tissue Culture Infectious Dose (TCID50)
2.4. Immunoblotting
2.5. Flow Cytometric Analysis
2.6. In Vivo Tumour Growth
2.7. Statistical Analysis
3. Results
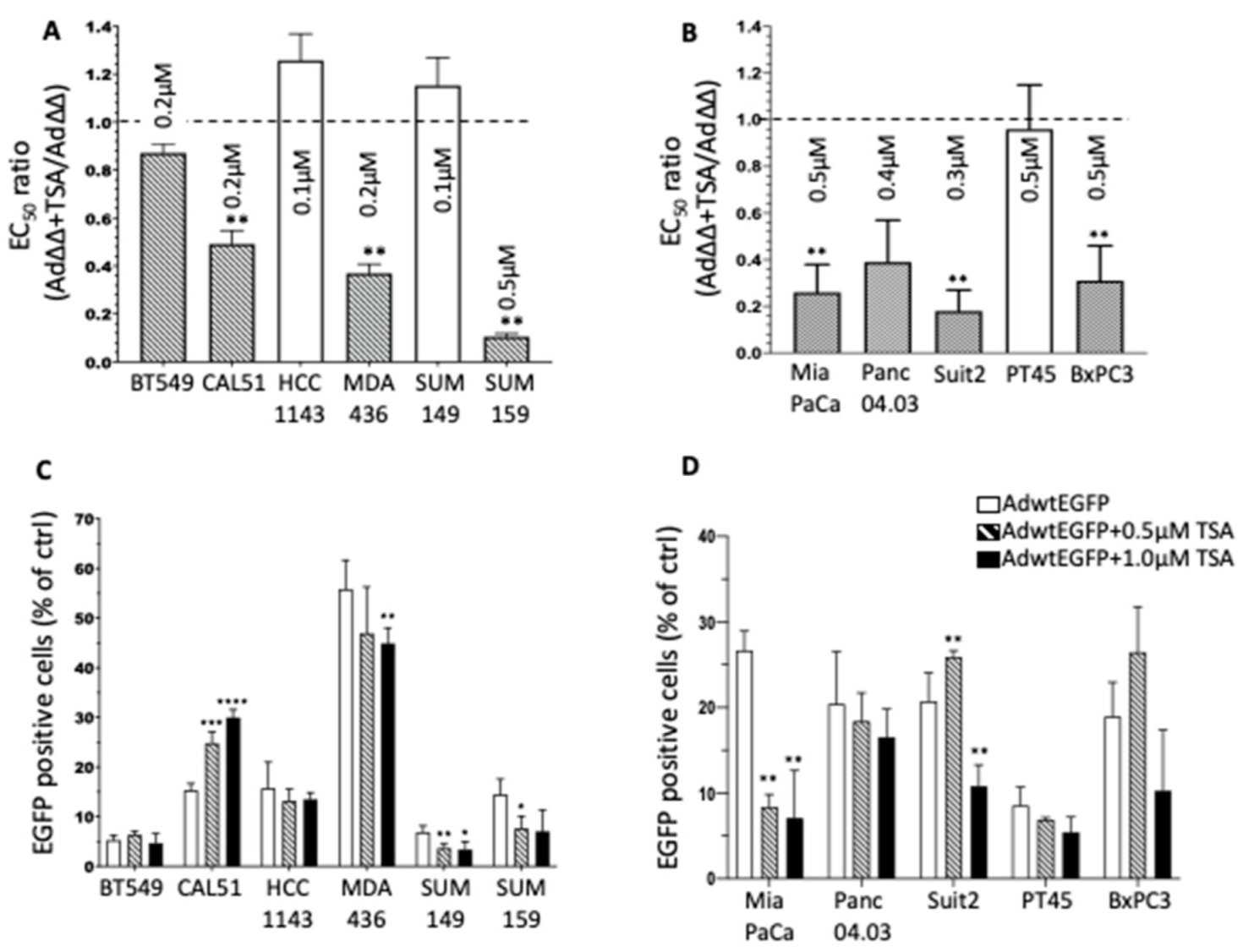
3.1. The Pan-HDACi TSA Sensitises TNBC and PDAC Cell Lines to Ad∆∆-Mediated Cell Killing
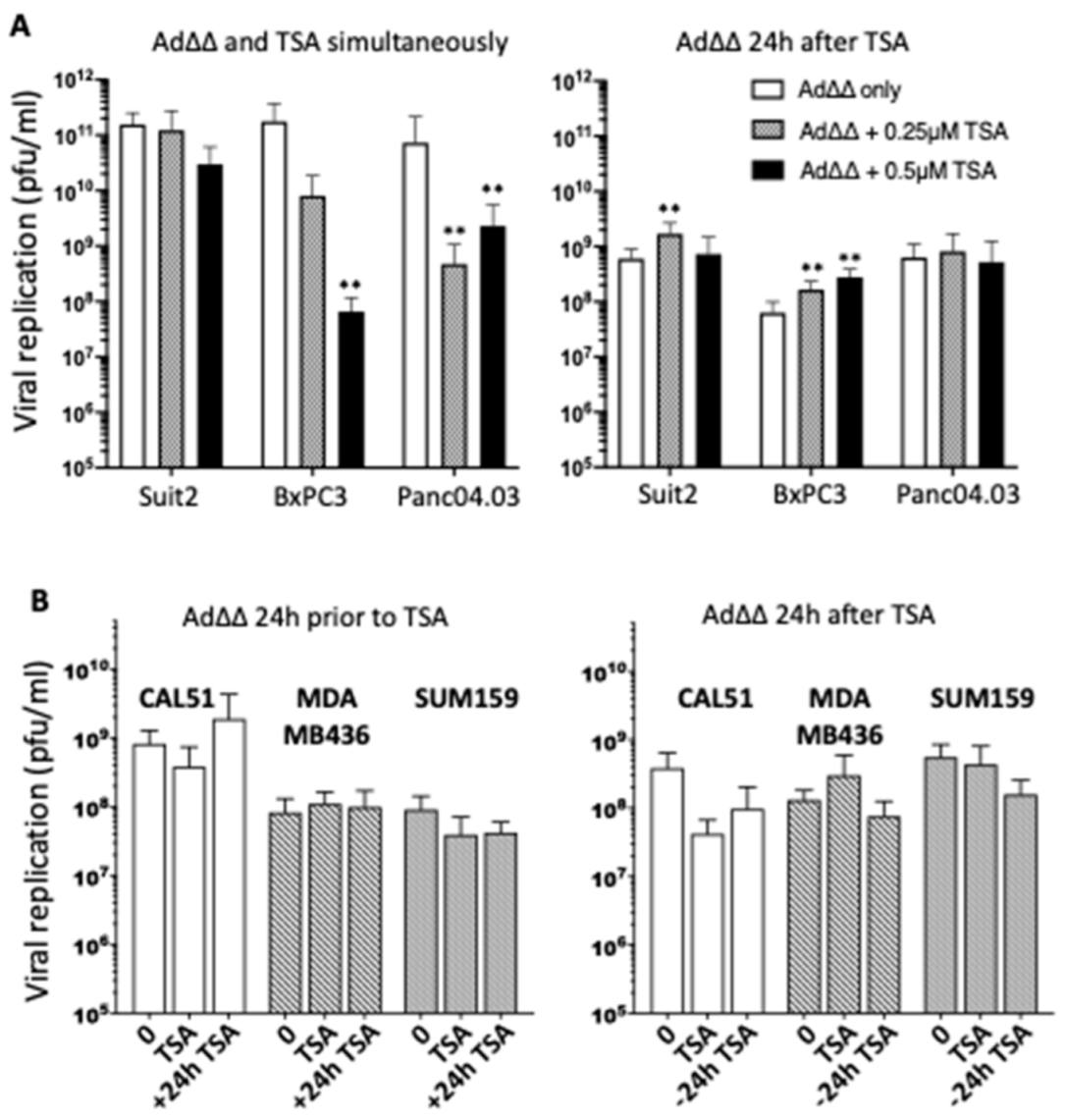
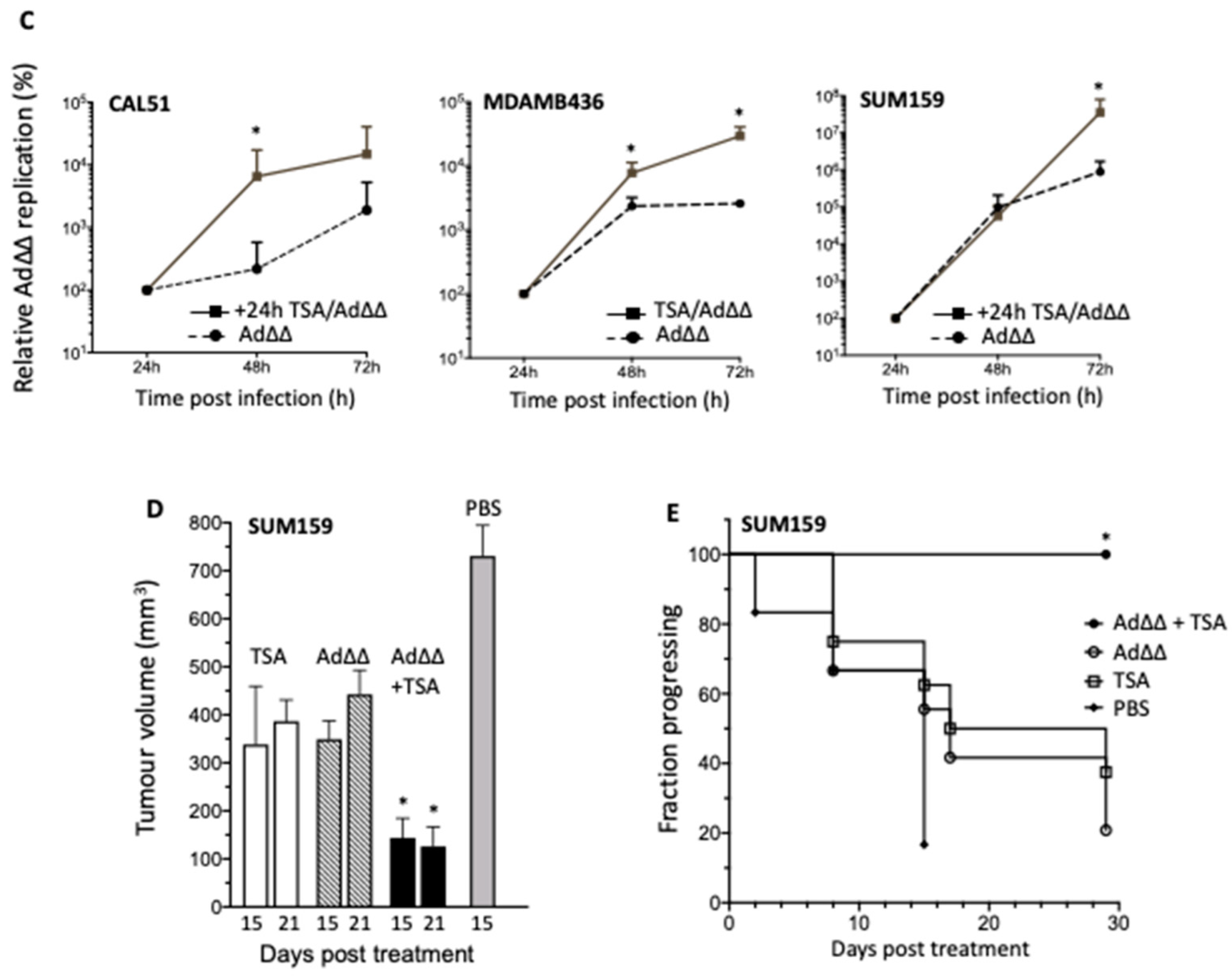
3.2. Effects on Viral Replication Is Dependent on Cell Line and Time of TSA Addition
3.3. TSA Promote Ad∆∆ Efficacy in SUM159 and Suit2 Tumour Xenografts In Vivo
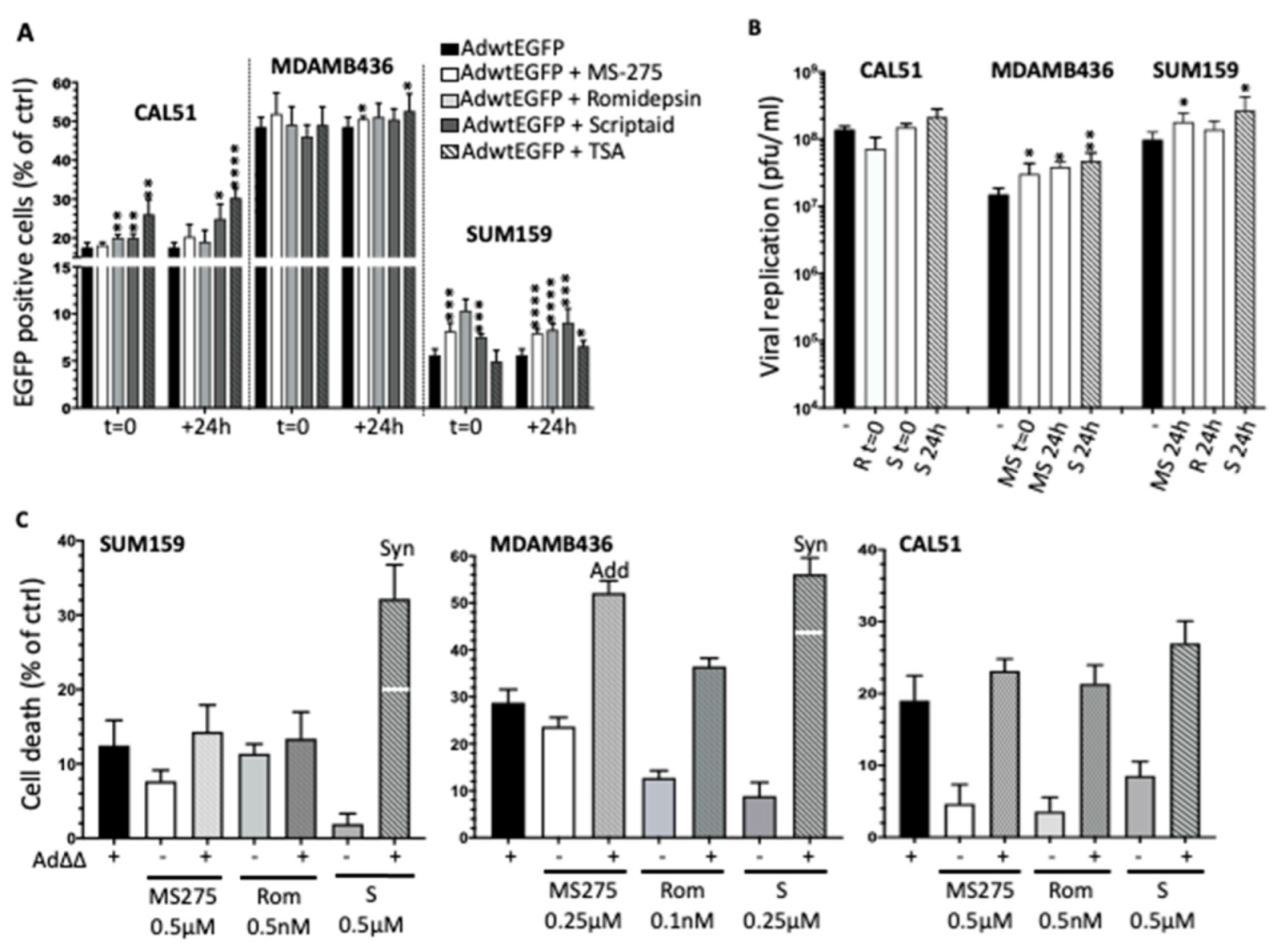
3.4. Selective HDAC Inhibitors Promote Ad∆∆ Infection and Propagation
3.5. Combination Treatments with Ad∆∆ and Scriptaid Enhance Virus-Induced Cell Death and Replication in TNBC Cell Lines
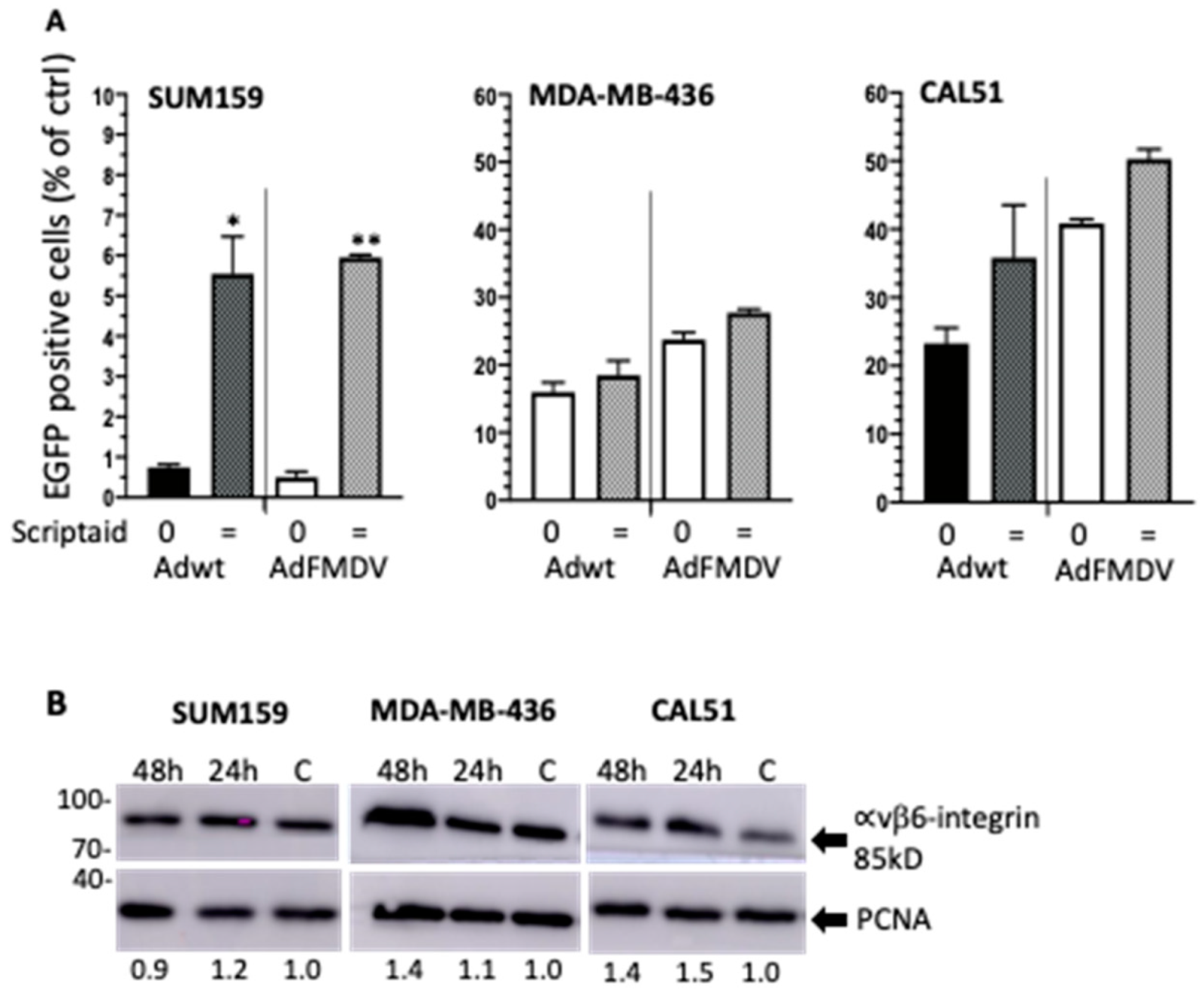
3.6. Infectivity of the αvß6-Integrin Targeted Mutant Ad5-3∆-A20T Is Enhanced by Scriptaid
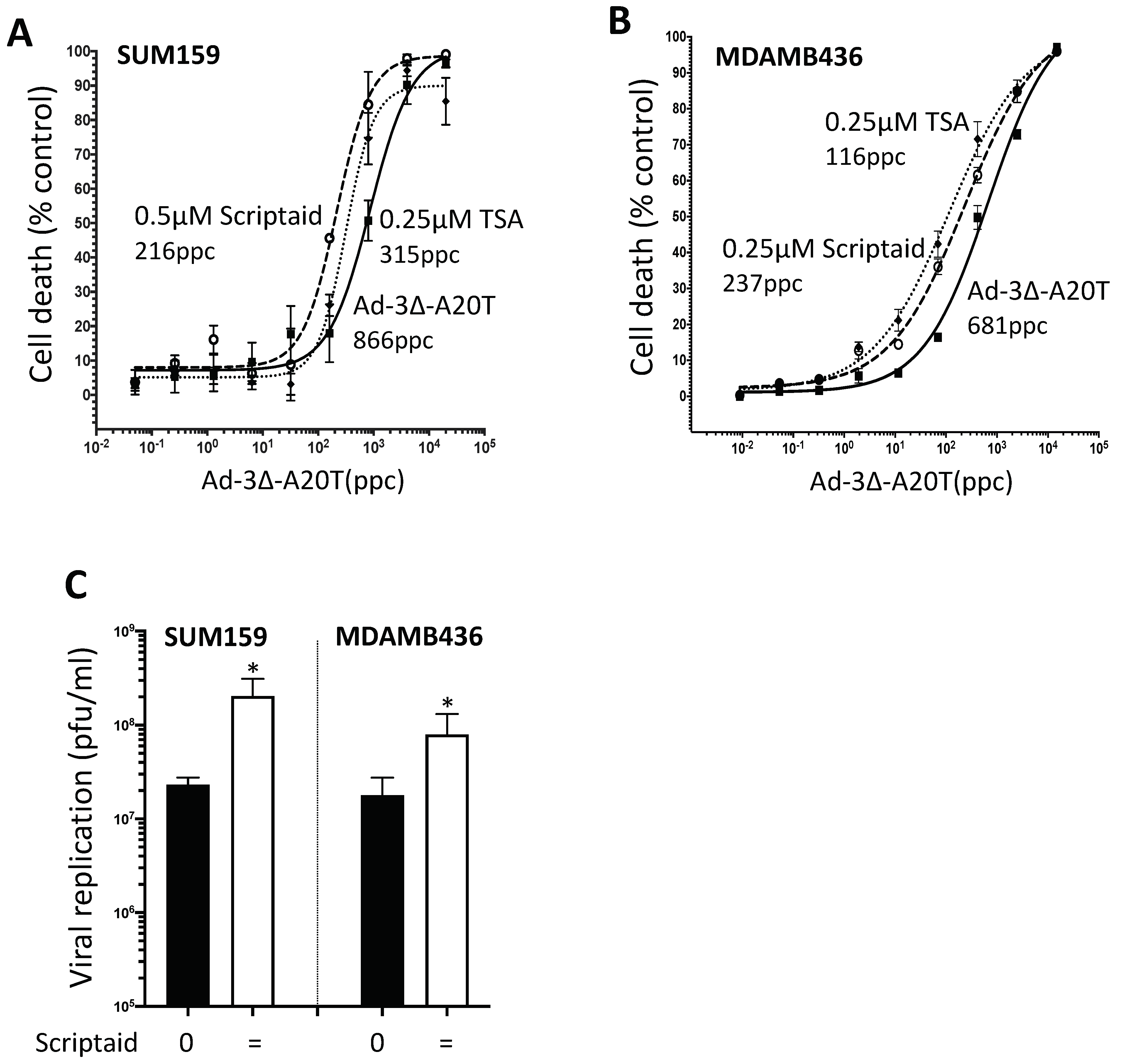
3.7. Scriptaid and TSA Enhance Ad5-3∆-A20T-Dependent Cell Killing and Replication in TNBC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wahba, H.A.; El-Hadaad, H.A. Current approaches in treatment of triple-negative breast cancer. Cancer Biol. Med. 2015, 12, 106–116. [Google Scholar] [CrossRef] [PubMed]
- Fietkau, R.; Grützmann, R.; Wittel, U.A.; Croner, R.S.; Jacobasch, L.; Neumann, U.P.; Reinacher-Schick, A.; Imhoff, D.; Boeck, S.; Keilholz, L.; et al. R0 resection following chemo (radio)therapy improves survival of primary inoperable pancreatic cancer patients. Interim results of the German randomized CONKO-007± trial. Strahlenther. Onkol. 2020, 197, 8–18. [Google Scholar] [CrossRef] [PubMed]
- Ingram, M.A.; Lauren, B.N.; Pumpalova, Y.; Park, J.; Lim, F.; Bates, S.E.; Kastrinos, F.; Manji, G.A.; Kong, C.Y.; Hur, C. Cost-effectiveness of neoadjuvant FOLFIRINOX versus gemcitabine plus nab-paclitaxel in borderline resectable/locally advanced pancreatic cancer patients. Cancer Rep. 2022, e1565. [Google Scholar] [CrossRef] [PubMed]
- Oettle, H. Progress in the knowledge and treatment of advanced pancreatic cancer: From benchside to bedside. Cancer Treat. Rev. 2014, 40, 1039–1047. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.R.; Suzuki, M. Immunology of Adenoviral Vectors in Cancer Therapy. Mol. Ther.-Methods Clin. Dev. 2019, 15, 418–429. [Google Scholar] [CrossRef] [PubMed]
- Öberg, D.; Yanover, E.; Adam, V.; Sweeney, K.; Costas, C.; Lemoine, N.; Halldén, G. Improved Potency and Selectivity of an Oncolytic E1ACR2 and E1B19K Deleted Adenoviral Mutant in Prostate and Pancreatic Cancers. Clin. Cancer Res. 2010, 16, 541–553. [Google Scholar] [CrossRef]
- Cherubini, G.; Kallin, C.; Mozetic, A.; Hammaren-Busch, K.; Muller, H.; Lemoine, N.R.; Hallden, G. The oncolytic adenovirus AdDeltaDelta enhances selective cancer cell killing in combination with DNA-damaging drugs in pancreatic cancer models. Gene Ther. 2011, 18, 1157–1165. [Google Scholar] [CrossRef]
- Pantelidou, C.; Cherubini, G.; Lemoine, N.R.; Hallden, G. The E1B19K-deleted oncolytic adenovirus mutant AdDelta19K sensitizes pancreatic cancer cells to drug-induced DNA-damage by down-regulating Claspin and Mre11. Oncotarget 2016, 7, 15703–15724. [Google Scholar] [CrossRef]
- Man, Y.K.S.; Davies, J.A.; Coughlan, L.; Pantelidou, C.; Blazquez-Moreno, A.; Marshall, J.F.; Parker, A.L.; Hallden, G. The Novel Oncolytic Adenoviral Mutant Ad5-3Delta-A20T Retargeted to alphavbeta6 Integrins Efficiently Eliminates Pancreatic Cancer Cells. Mol. Cancer Ther. 2018, 17, 575–587. [Google Scholar] [CrossRef]
- Man, Y.K.S.; Foster, J.; Carapuça, E.; Davies, J.; Parker, A.L.; Sosabowski, J.; Halldén, G. Systemic delivery and SPECT/CT in vivo imaging of 125I-labelled oncolytic adenoviral mutants in models of pancreatic cancer. Sci. Rep. 2019, 9, 12840. [Google Scholar] [CrossRef]
- Leitner, S.; Sweeney, K.; Öberg, D.; Davies, D.; Miranda, E.; Lemoine, N.R.; Halldén, G. Oncolytic Adenoviral Mutants with E1B19K Gene Deletions Enhance Gemcitabine-induced Apoptosis in Pancreatic Carcinoma Cells and Anti-Tumor Efficacy In vivo. Clin. Cancer Res. 2009, 15, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Aguirre-Hernández, C.; Maya-Pineda, H.; Millán, J.S.; Man, Y.K.S.; Lu, Y.J.; Halldén, G. Sensitisation to mitoxantrone-induced apoptosis by the oncolytic adenovirus Ad∆∆ through Bcl-2-dependent attenuation of autophagy. Oncogenesis 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Coughlan, L.; Vallath, S.; Gros, A.; Gimenez-Alejandre, M.; van Rooijen, N.; Thomas, G.J.; Baker, A.H.; Cascallo, M.; Alemany, R.; Hart, I.R. Combined fiber modifications both to target alpha(v)beta(6) and detarget the coxsackievirus-adenovirus receptor improve virus toxicity profiles in vivo but fail to improve antitumoral efficacy relative to adenovirus serotype 5. Hum. Gene Ther. 2012, 23, 960–979. [Google Scholar] [CrossRef] [PubMed]
- Desai, K.; Nair, M.G.; Prabhu, J.S.; Vinod, A.; Korlimarla, A.; Rajarajan, S.; Aiyappa, R.; Kaluve, R.S.; Alexander, A.; Hari, P.S.; et al. High expression of integrin beta6 in association with the Rho-Rac pathway identifies a poor prognostic subgroup within HER2 amplified breast cancers. Cancer Med. 2016, 5, 2000–2011. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.D.; Thomas, G.J.; Clark, S.; Dawoud, M.M.; Vallath, S.; Payne, S.J.; Gomm, J.J.; Dreger, S.A.; Dickinson, S.; Edwards, D.R.; et al. Altered microenvironment promotes progression of preinvasive breast cancer: Myoepithelial expression of alphavbeta6 integrin in DCIS identifies high-risk patients and predicts recurrence. Clin. Cancer Res. 2014, 20, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Hsu, E.; Pennella, M.A.; Zemke, N.R.; Eng, C.; Berk, A.J. Adenovirus E1A Activation Domain Regulates H3 Acetylation Affecting Varied Steps in Transcription at Different Viral Promoters. J. Virol. 2018, 92, e00805-18. [Google Scholar] [CrossRef] [PubMed]
- Zemke, N.R.; Gou, D.; Berk, A.J. Dedifferentiation by adenovirus E1A due to inactivation of Hippo pathway effectors YAP and TAZ. Genes Dev. 2019, 33, 828–843. [Google Scholar] [CrossRef]
- Ferrari, R.; Gou, D.; Jawdekar, G.; Johnson, S.A.; Nava, M.; Su, T.; Yousef, A.F.; Zemke, N.R.; Pellegrini, M.; Kurdistani, S.K.; et al. Adenovirus small E1A employs the lysine acetylases p300/CBP and tumor suppressor Rb to repress select host genes and promote productive virus infection. Cell Host Microbe 2014, 16, 663–676. [Google Scholar] [CrossRef]
- Ferrari, R.; Su, T.; Li, B.; Bonora, G.; Oberai, A.; Chan, Y.; Sasidharan, R.; Berk, A.J.; Pellegrini, M.; Kurdistani, S.K. Reorganization of the host epigenome by a viral oncogene. Genome Res. 2012, 22, 1212–1221. [Google Scholar] [CrossRef]
- Wang, Z.; Zang, C.; Cui, K.; Schones, D.E.; Barski, A.; Peng, W.; Zhao, K. Genome-wide Mapping of HATs and HDACs Reveals Distinct Functions in Active and Inactive Genes. Cell 2009, 138, 1019–1031. [Google Scholar] [CrossRef]
- Chiocca, E.A.; Nakashima, H.; Nguyen, T. Combining HDAC inhibitors with oncolytic virotherapy for cancer therapy. Oncolytic Virother. 2015, 4, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Miquel-Cases, A.; Retèl, V.P.; van Harten, W.H.; Steuten, L.M. Decisions on Further Research for Predictive Biomarkers of High-Dose Alkylating Chemotherapy in Triple-Negative Breast Cancer: A Value of Information Analysis. Value Health 2016, 19, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Loo, S.Y.; Syn, N.L.; Koh, A.P.; Teng, J.C.; Deivasigamani, A.; Tan, T.Z.; Thike, A.A.; Vali, S.; Kapoor, S.; Wang, X.; et al. Epigenetic derepression converts PPARgamma into a druggable target in triple-negative and endocrine-resistant breast cancers. Cell Death Discov. 2021, 7, 265. [Google Scholar] [CrossRef] [PubMed]
- Eckschlager, T.; Plch, J.; Stiborova, M.; Hrabeta, J. Histone Deacetylase Inhibitors as Anticancer Drugs. Int. J. Mol. Sci. 2017, 18, 1414. [Google Scholar] [CrossRef]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, Y.; Negishi, H.; Idogawa, M.; Suzuki, H.; Mita, H.; Toyota, M.; Shinomura, Y.; Imai, K.; Tokino, T. Histone deacetylase inhibitor FK228 enhances adenovirus-mediated p53 family gene therapy in cancer models. Mol. Cancer Ther. 2008, 7, 779–787. [Google Scholar] [CrossRef][Green Version]
- Goldsmith, M.E.; Aguila, A.; Steadman, K.; Martinez, A.; Steinberg, S.M.; Alley, M.C.; Waud, W.R.; Bates, S.E.; Fojo, T. The histone deacetylase inhibitor FK228 given prior to adenovirus infection can boost infection in melanoma xenograft model systems. Mol. Cancer Ther. 2007, 6, 496–505. [Google Scholar] [CrossRef][Green Version]
- Berghauser Pont, L.M.; Kleijn, A.; Kloezeman, J.J.; van den Bossche, W.; Kaufmann, J.K.; de Vrij, J.; Leenstra, S.; Dirven, C.M.; Lamfers, M.L. The HDAC Inhibitors Scriptaid and LBH589 Combined with the Oncolytic Virus Delta24-RGD Exert Enhanced Anti-Tumor Efficacy in Patient-Derived Glioblastoma Cells. PLoS ONE 2015, 10, e0127058. [Google Scholar] [CrossRef]
- Hulin-Curtis, S.L.; Davies, J.A.; Jones, R.; Hudson, E.; Hanna, L.; Chester, J.D.; Parker, A.L. Histone deacetylase inhibitor trichostatin A sensitises cisplatin-resistant ovarian cancer cells to oncolytic adenovirus. Oncotarget 2018, 9, 26328–26341. [Google Scholar] [CrossRef][Green Version]
- Lee, C.T.; Kim, D.R.; Park, M.Y.; Lim, H.J.; Park, J.S.; Cho, Y.J.; Lee, S.W.; Yoon, H.I.; Lee, J.H.; Kim, Y.S. Combination therapy of conditionally replicating adenovirus and histone deacetylase inhibitors. Int. J. Mol. Med. 2011, 29, 218–224. [Google Scholar] [CrossRef]
- Marks, P.A. Histone deacetylase inhibitors: A chemical genetics approach to understanding cellular functions. Biochim. Biophys. Acta 2010, 1799, 717–725. [Google Scholar] [CrossRef] [PubMed]
- Takai, N.; Ueda, T.; Nishida, M.; Nasu, K.; Narahara, H. A novel histone deacetylase inhibitor, Scriptaid, induces growth inhibition, cell cycle arrest and apoptosis in human endometrial cancer and ovarian cancer cells. Int. J. Mol. Med. 2006, 17, 323–329. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cody, J.J.; Hurst, D.R. Promising oncolytic agents for metastatic breast cancer treatment. Oncolytic Virother. 2015, 4, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Nattress, C.B.; Halldén, G. Advances in oncolytic adenovirus therapy for pancreatic cancer. Cancer Lett. 2018, 434, 56–69. [Google Scholar] [CrossRef]
- Bazan-Peregrino, M.; Garcia-Carbonero, R.; Laquente, B.; Álvarez, R.; Mato-Berciano, A.; Gimenez-Alejandre, M.; Morgado, S.; Rodríguez-García, A.; Maliandi, M.V.; Riesco, M.C.; et al. VCN-01 disrupts pancreatic cancer stroma and exerts antitumor effects. J. Immunother. Cancer 2021, 9, e003254. [Google Scholar] [CrossRef]
- Gao, J.; Zhang, W.; Ehrhardt, A. Expanding the Spectrum of Adenoviral Vectors for Cancer Therapy. Cancers 2020, 12, 1139. [Google Scholar] [CrossRef]
- Hauschild, A.; Trefzer, U.; Garbe, C.; Kaehler, K.C.; Ugurel, S.; Kiecker, F.; Eigentler, T.; Krissel, H.; Schott, A.; Schadendorf, D. Multicenter phase II trial of the histone deacetylase inhibitor pyridylmethyl-N-{4-[(2-aminophenyl)-carbamoyl]-benzyl}-carbamate in pretreated metastatic melanoma. Melanoma Res. 2008, 18, 274–278. [Google Scholar] [CrossRef]
- Gojo, I.; Jiemjit, A.; Trepel, J.B.; Sparreboom, A.; Figg, W.D.; Rollins, S.; Tidwell, M.L.; Greer, J.; Chung, E.J.; Lee, M.J.; et al. Phase 1 and pharmacologic study of MS-275, a histone deacetylase inhibitor, in adults with refractory and relapsed acute leukemias. Blood 2006, 109, 2781–2790. [Google Scholar] [CrossRef]
- Sachs, M.D.; Ramamurthy, M.; Van Der Poel, H.; Wickham, T.J.; Lamfers, M.; Gerritsen, W.; Chowdhury, W.; Li, Y.; Schoenberg, M.P.; Rodriguez, R. Histone deacetylase inhibitors upregulate expression of the coxsackie adenovirus receptor (CAR) preferentially in bladder cancer cells. Cancer Gene Ther. 2004, 11, 477–486. [Google Scholar] [CrossRef]
- Liu, L.; Sun, X.; Xie, Y.; Zhuang, Y.; Yao, R.; Xu, K. Anticancer effect of histone deacetylase inhibitor scriptaid as a single agent for hepatocellular carcinoma. Biosci. Rep. 2018, 38, BSR20180360. [Google Scholar] [CrossRef]
- Bressy, C.; Majhen, D.; Raddi, N.; Jdey, W.; Cornilleau, G.; Zig, L.; Guirouilh-Barbat, J.; Lopez, B.; Bawa, O.; Opolon, P.; et al. Combined therapy of colon carcinomas with an oncolytic adenovirus and valproic acid. Oncotarget 2017, 8, 97344–97360. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, J.; Bonifati, S.; Hristov, G.; Marttila, T.; Valmary-Degano, S.; Stanzel, S.; Schnölzer, M.; Mougin, C.; Aprahamian, M.; Grekova, S.P.; et al. Synergistic combination of valproic acid and oncolytic parvovirus H-1 PV as a potential therapy against cervical and pancreatic carcinomas. EMBO Mol. Med. 2013, 5, 1537–1555. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Nakajima, S.I.; Niizeki, H.; Tada, M.; Nakagawa, K.; Kondo, S.; Okada, F. Trichostatin A with adenovirus-mediated p53 gene transfer synergistically induces apoptosis in breast cancer cell line MDA-MB-231. Oncol. Rep. 2009, 22, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Okegawa, T.; Sayne, J.R.; Nutahara, K.; Pong, R.C.; Saboorian, H.; Kabbani, W.; Higashihara, E.; Hsieh, J.T. A Histone Deacetylase Inhibitor Enhances Adenoviral Infection of Renal Cancer Cells. J. Urol. 2007, 177, 1148–1156. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, T.; Hioki, M.; Fujiwara, T.; Nishizaki, M.; Kagawa, S.; Taki, M.; Kishimoto, H.; Endo, Y.; Urata, Y.; Tanaka, N. Histone deacetylase inhibitor FR901228 enhances the antitumor effect of telomerase-specific replication-selective adenoviral agent OBP-301 in human lung cancer cells. Exp. Cell Res. 2006, 312, 256–265. [Google Scholar] [CrossRef] [PubMed]
- Saha, B.; Parks, R.J. Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Suppresses Human Adenovirus Gene Expression and Replication. J. Virol. 2019, 93, e00088-19. [Google Scholar] [CrossRef] [PubMed]
- Stern, H.M.; Gardner, H.; Burzykowski, T.; Elatre, W.; O’Brien, C.; Lackner, M.; Pestano, G.A.; Santiago, A.; Villalobos, I.; Eiermann, W.; et al. PTEN Loss Is Associated with Worse Outcome in HER2-Amplified Breast Cancer Patients but Is not Associated with Trastuzumab Resistance. Clin. Cancer Res. 2015, 21, 2065–2074. [Google Scholar] [CrossRef]
- Barnabas, N.; Cohen, D. Phenotypic and Molecular Characterization of MCF10DCIS and SUM Breast Cancer Cell Lines. Int. J. Breast Cancer 2013, 2013, 872743. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rodríguez, M.D.C.R.; Rodríguez, I.G.; Nattress, C.; Qureshi, A.; Halldén, G. HDAC Inhibitors Enhance Efficacy of the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T in Pancreatic and Triple-Negative Breast Cancer Models. Viruses 2022, 14, 1006. https://doi.org/10.3390/v14051006
Rodríguez MDCR, Rodríguez IG, Nattress C, Qureshi A, Halldén G. HDAC Inhibitors Enhance Efficacy of the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T in Pancreatic and Triple-Negative Breast Cancer Models. Viruses. 2022; 14(5):1006. https://doi.org/10.3390/v14051006
Chicago/Turabian StyleRodríguez, María Del Carmen Rodríguez, Inés García Rodríguez, Callum Nattress, Ahad Qureshi, and Gunnel Halldén. 2022. "HDAC Inhibitors Enhance Efficacy of the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T in Pancreatic and Triple-Negative Breast Cancer Models" Viruses 14, no. 5: 1006. https://doi.org/10.3390/v14051006
APA StyleRodríguez, M. D. C. R., Rodríguez, I. G., Nattress, C., Qureshi, A., & Halldén, G. (2022). HDAC Inhibitors Enhance Efficacy of the Oncolytic Adenoviruses Ad∆∆ and Ad-3∆-A20T in Pancreatic and Triple-Negative Breast Cancer Models. Viruses, 14(5), 1006. https://doi.org/10.3390/v14051006