
Genetic Characterization of Small Ruminant Lentiviruses (SRLVs) Circulating in Naturally Infected Sheep in Central Italy

 ,
,  , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. SRLV Samples Collection and DNA Extraction
2.2. SRLV Proviral Amplification and Bioinformatic Analysis
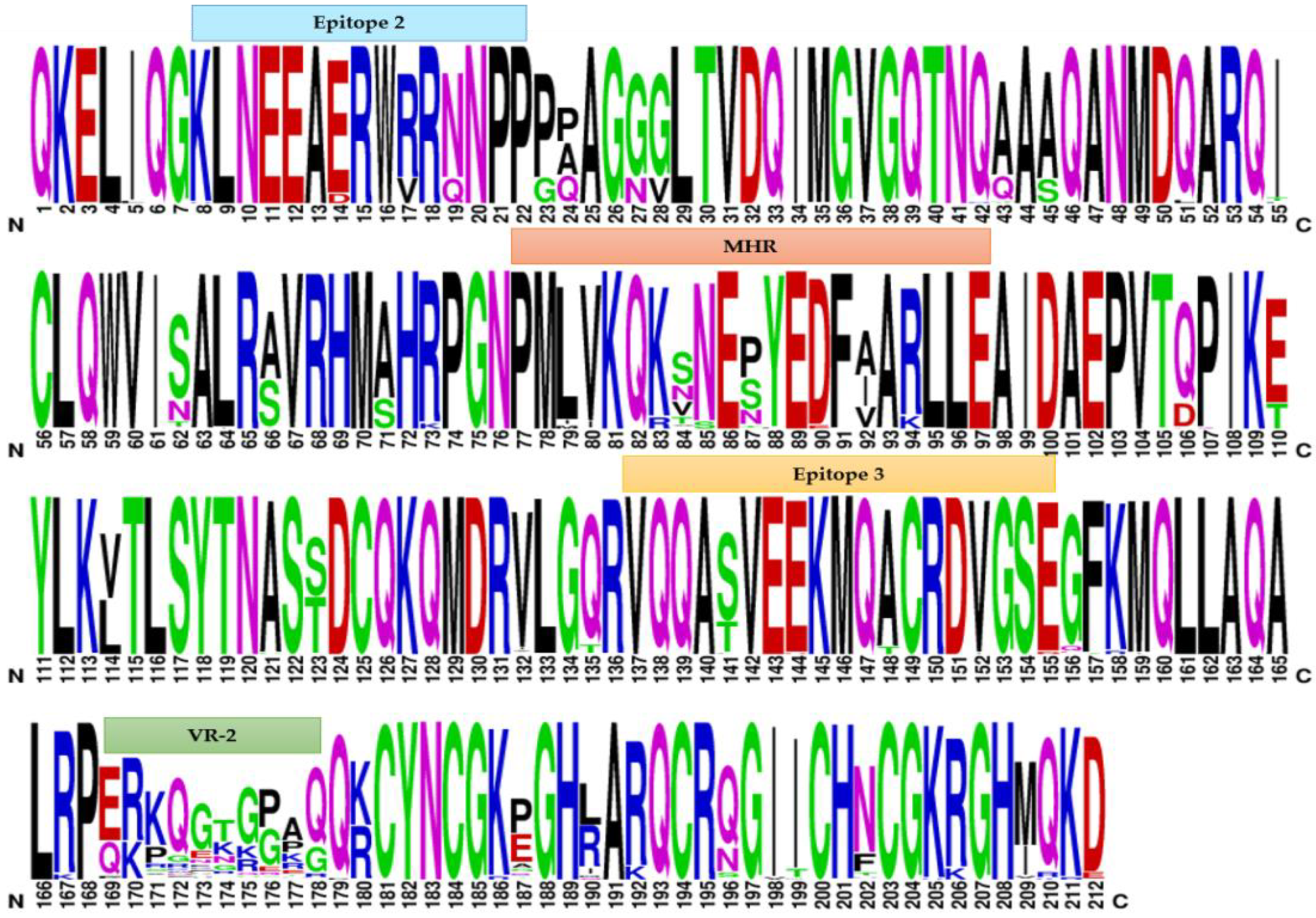
2.3. Weblogo Diagrams
2.4. Estimation of Nonsynonymous and Synonymous Substitution Rates
3. Results
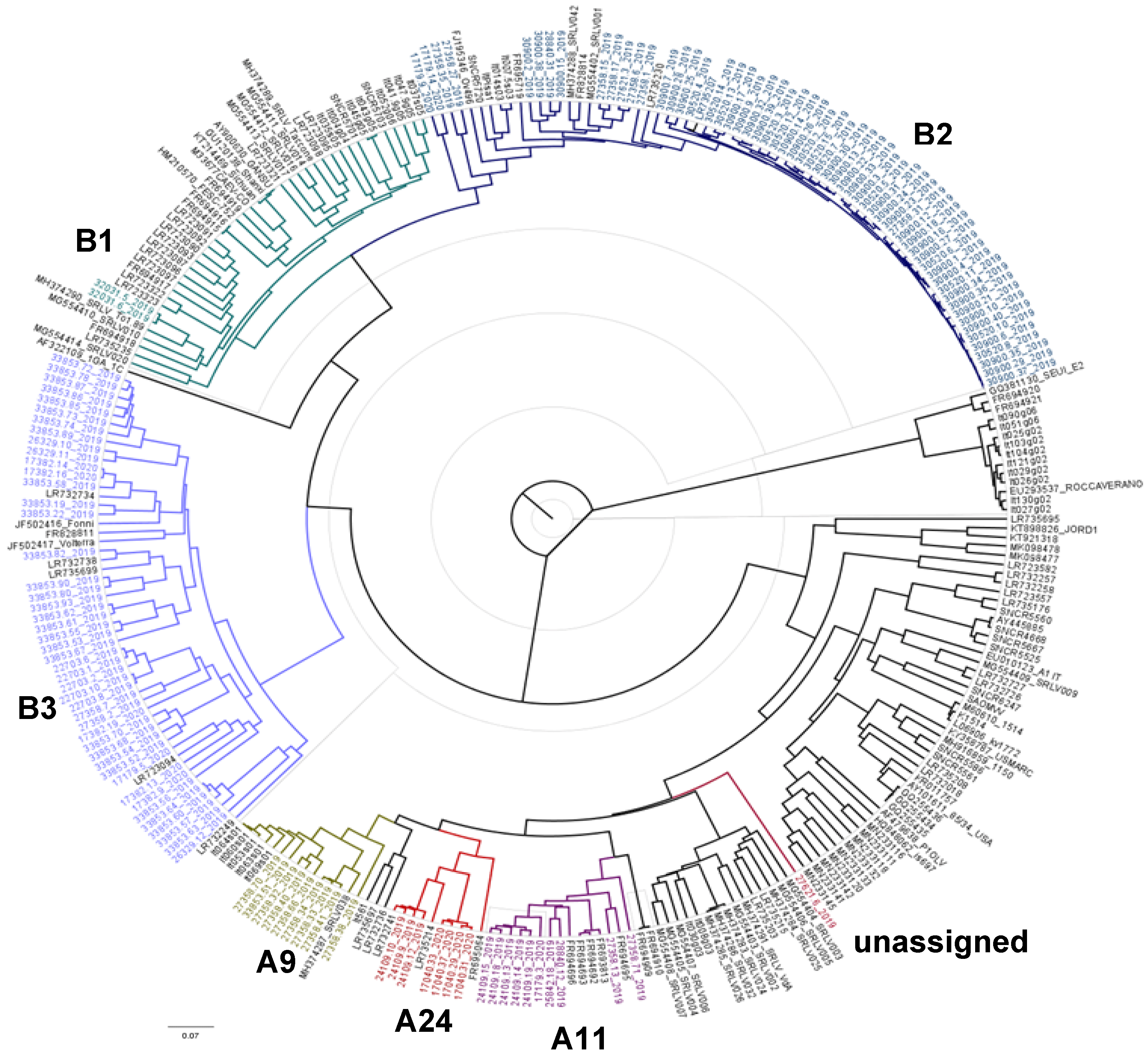
3.1. Phylogenetic Analysis
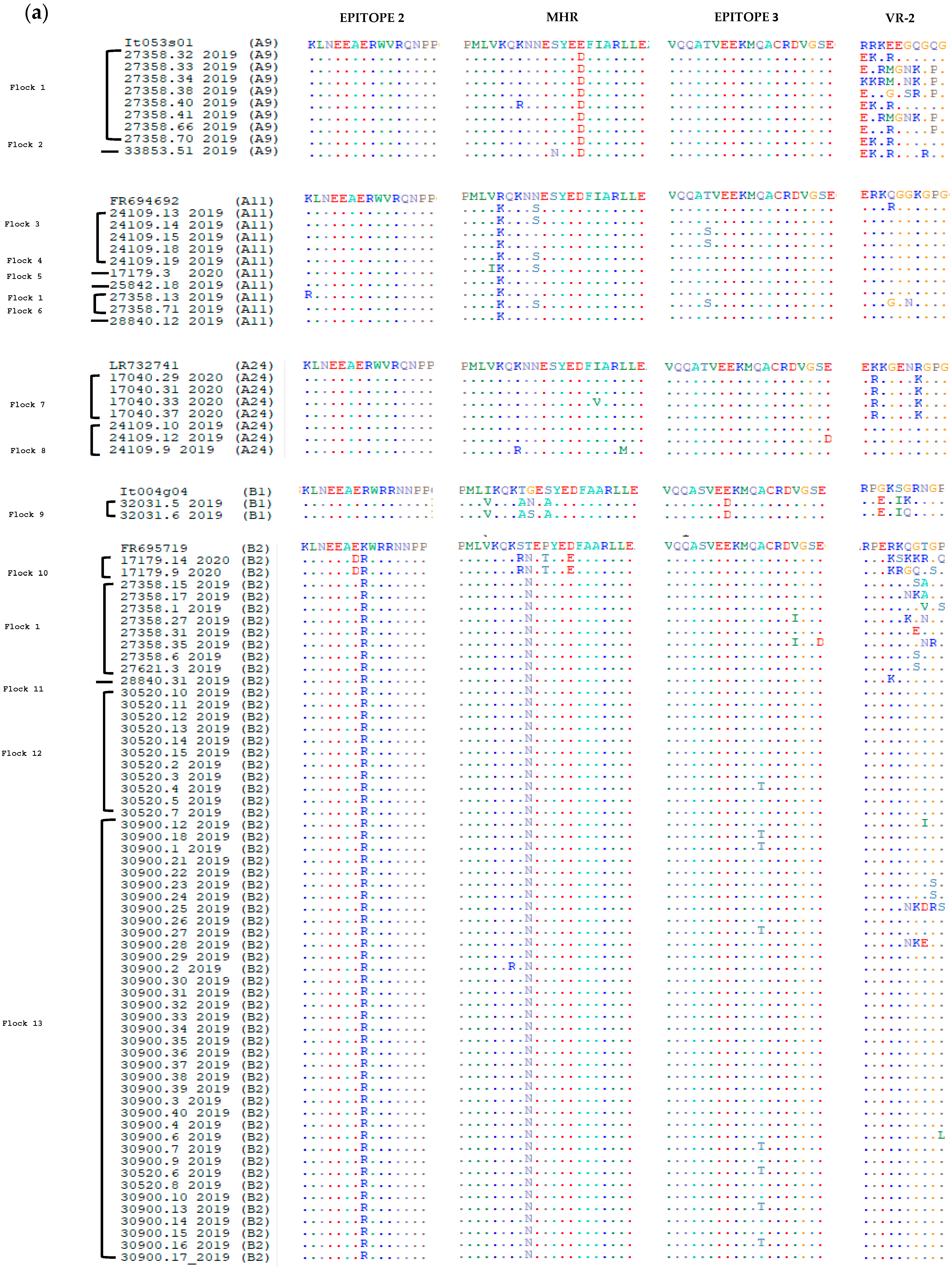
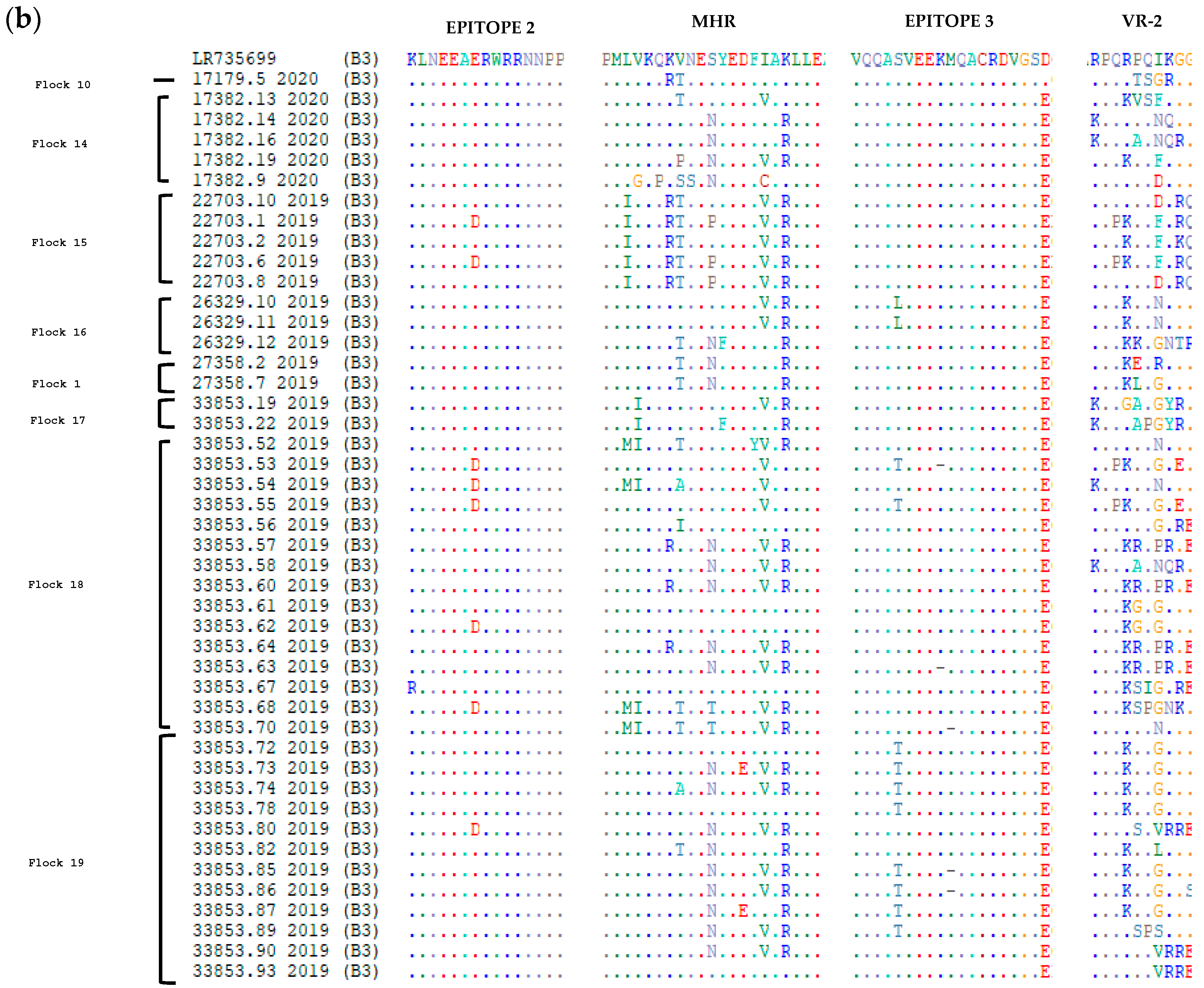
3.2. Comparative Analysis of Immunodominant Regions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Krupovic, M.; Blomberg, J.; Coffin, J.M.; Dasgupta, I.; Fan, H.; Geering, A.D.; Gifford, R.; Harrach, B.; Hull, R.; Johnson, W.; et al. Ortervirales: New Virus Order Unifying Five Families of Reverse-Transcribing Viruses. J. Virol. 2018, 92, e00515-18. [Google Scholar] [CrossRef] [PubMed]
- Kalogianni, A.I.; Bossis, I.; Ekateriniadou, L.V.; Gelasakis, A.I. Etiology, Epizootiology and Control of Maedi-Visna in Dairy Sheep: A Review. Animals 2020, 10, 616. [Google Scholar] [CrossRef] [PubMed]
- Santry, L.A.; de Jong, J.; Gold, A.C.; Walsh, S.R.; Menzies, P.I.; Wootton, S.K. Genetic characterization of small ruminant lentiviruses circulating in naturally infected sheep and goats in Ontario, Canada. Virus Res. 2013, 175, 30–44. [Google Scholar] [CrossRef]
- De Pablo-Maiso, L.; Echeverría, I.; Rius-Rocabert, S.; Luján, L.; Garcin, D.; de Andrés, D.; Nistal-Villán, E.; Reina, R. Sendai Virus, a Strong Inducer of Anti-Lentiviral State in Ovine Cells. Vaccines 2020, 8, 206. [Google Scholar] [CrossRef] [PubMed]
- Blacklaws, B.A. Small ruminant lentiviruses: Immunopathogenesis of visna-maedi and caprine arthritis and encephalitis virus. Comp. Immunol. Microbiol. Infect. Dis. 2012, 35, 259–269. [Google Scholar] [CrossRef]
- Minguijón, E.; Reina, R.; Pérez, M.; Polledo, L.; Villoria, M.; Ramírez, H.; Leginagoikoa, I.; Badiola, J.J.; García-Marín, J.F.; de Andrés, D.; et al. Small ruminant lentivirus infections and diseases. Vet. Microbiol. 2015, 181, 75–89. [Google Scholar] [CrossRef]
- Acevedo Jiménez, G.E.; Tórtora Pérez, J.L.; Rodríguez Murillo, C.; Arellano Reynoso, B.; Ramírez Álvarez, H. Serotyping versus genotyping in infected sheep and goats with small ruminant lentiviruses. Vet. Microbiol. 2020, 252, 108931. [Google Scholar] [CrossRef]
- Furtado Araújo, J.; Andrioli, A.; Pinheiro, R.R.; Sider, L.H.; de Sousa, A.L.M.; de Azevedo, D.A.A.; Peixoto, R.M.; Lima, A.M.C.; Damasceno, E.M.; Souza, S.C.R.; et al. Vertical transmissibility of small ruminant lentivirus. PLoS ONE 2020, 15, e0239916. [Google Scholar] [CrossRef]
- De Miguel, R.; Arrieta, M.; Rodríguez-Largo, A.; Echeverría, I.; Resendiz, R.; Pérez, E.; Ruiz, H.; Pérez, M.; de Andrés, D.; Reina, R.; et al. Worldwide Prevalence of Small Ruminant Lentiviruses in Sheep: A Systematic Review and Meta-Analysis. Animals 2021, 11, 784. [Google Scholar] [CrossRef]
- Nowicka, D.; Czopowicz, M.; Mickiewicz, M.; Szaluś-Jordanow, O.; Witkowski, L.; Bagnicka, E.; Kaba, J. Diagnostic performance of ID screen MVV-CAEV Indirect Screening ELISA in identifying small ruminant lentiviruses-infected goats. Pol. J. Vet. Sci. 2014, 17, 501–506. [Google Scholar] [CrossRef][Green Version]
- Ramírez, H.; Reina, R.; Amorena, B.; Andrés, D.D.; Martínez, H.A. Small Ruminant Lentiviruses: Genetic Variability, Tropism and Diagnosis. Viruses 2013, 5, 1175–1207. [Google Scholar] [CrossRef] [PubMed]
- Pépin, M.; Vitu, C.; Russo, P.; Mornex, J.F.; Peterhans, E. Maedi-visna virus infection in sheep: A review. Vet. Res. 1998, 29, 341–367. [Google Scholar]
- Gomez-Lucia, E.; Barquero, N.; Domenech, A. Maedi-Visna virus: Current perspectives. Vet. Med. Res. Rep. 2018, 9, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Querat, G.; Audoly, G.; Sonigo, P.; Vigne, R. Nucleotide sequence analysis of SA-OMVV, a visna-related ovine lentivirus: Phylogenetic history of lentiviruses. Virology 1990, 175, 434–447. [Google Scholar] [CrossRef]
- Vigne, R.; Filippi, P.; Quérat, G.; Sauze, N.; Vitu, C.; Russo, P.; Delori, P. Precursor Polypeptides to Structural Proteins of Visna Virus. J. Virol. 1982, 42, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Olech, M.; Valas, S.; Kuźmak, J. Epidemiological survey in single-species flocks from Poland reveals expanded genetic and antigenic diversity of small ruminant lentiviruses. PLoS ONE 2018, 13, e0193892. [Google Scholar] [CrossRef] [PubMed]
- Wainhobson, S. Running the gamut of retroviral variation. Trends Microbiol. 1996, 4, 135–141. [Google Scholar] [CrossRef]
- Olech, M.; Kuźmak, J. Molecular Characterization of Small Ruminant Lentiviruses of Subtype A5 Detected in Naturally Infected but Clinically Healthy Goats of Carpathian Breed. Pathogens 2020, 9, 992. [Google Scholar] [CrossRef]
- Gayo, E.; Cuteri, V.; Polledo, L.; Rossi, G.; García Marín, J.; Preziuso, S. Genetic Characterization and Phylogenetic Analysis of Small Ruminant Lentiviruses Detected in Spanish Assaf Sheep with Different Mammary Lesions. Viruses 2018, 10, 315. [Google Scholar] [CrossRef]
- Urbanska, D.; Puchała, R.; Jarczak, J.; Czopowicz, M.; Kaba, J.; Horbańczuk, K.; Bagnicka, E. Does Small Ruminant Lentivirus Infection in Goats Predispose to Bacterial Infection of the Mammary Gland? A Preliminary Study. Animals 2021, 11, 1851. [Google Scholar] [CrossRef]
- Murphy, F.A. Veterinary Virology; Academic Press: San Diego, CA, USA, 1999; ISBN 9780080552033. [Google Scholar]
- Shah, C.; Böni, J.; Huder, J.B.; Vogt, H.-R.; Mühlherr, J.; Zanoni, R.; Miserez, R.; Lutz, H.; Schüpbach, J. Phylogenetic analysis and reclassification of caprine and ovine lentiviruses based on 104 new isolates: Evidence for regular sheep-to-goat transmission and worldwide propagation through livestock trade. Virology 2004, 319, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Peeters, M.; Esu-Williams, E.; Vergne, L.; Montavon, C.; Mulanga-Kabeya, C.; Harry, T.; Ibironke, A.; Lesage, D.; Patrel, D.; Delaporte, E. Predominance of subtype A and G HIV type 1 in Nigeria, with geographical differences in their distribution. AIDS Res. Hum. Retrovir. 2000, 16, 315–325. [Google Scholar] [CrossRef] [PubMed]
- Bazzucchi, M.; Pierini, I.; Gobbi, P.; Pirani, S.; Torresi, C.; Iscaro, C.; Feliziani, F.; Giammarioli, M. Genomic Epidemiology and Heterogeneity of SRLV in Italy from 1998 to 2019. Viruses 2021, 13, 2338. [Google Scholar] [CrossRef] [PubMed]
- Michiels, R.; Adjadj, N.R.; De Regge, N. Phylogenetic Analysis of Belgian Small Ruminant Lentiviruses Supports Cross Species Virus Transmission and Identifies New Subtype B5 Strains. Pathogens 2020, 9, 183. [Google Scholar] [CrossRef]
- Molaee, V.; Bazzucchi, M.; De Mia, G.M.; Otarod, V.; Abdollahi, D.; Rosati, S.; Lühken, G. Phylogenetic analysis of small ruminant lentiviruses in Germany and Iran suggests their expansion with domestic sheep. Sci. Rep. 2020, 10, 2243. [Google Scholar] [CrossRef]
- Bertolotti, L.; Mazzei, M.; Puggioni, G.; Carrozza, M.L.; Dei Giudici, S.; Muz, D.; Juganaru, M.; Patta, C.; Tolari, F.; Rosati, S. Characterization of new small ruminant lentivirus subtype B3 suggests animal trade within the Mediterranean Basin. J. Gen. Virol. 2011, 92, 1923–1929. [Google Scholar] [CrossRef]
- L’Homme, Y.; Leboeuf, A.; Arsenault, J.; Fras, M. Identification and Characterization of an Emerging Small Ruminant Lentivirus Circulating Recombinant Form (CRF). Virology 2015, 475, 159–171. [Google Scholar] [CrossRef]
- Gjerset, B.; Storset, A.K.; Rimstad, E. Genetic diversity of small-ruminant lentiviruses: Characterization of Norwegian isolates of Caprine arthritis encephalitis virus. J. Gen. Virol. 2006, 87, 573–580. [Google Scholar] [CrossRef]
- Giammarioli, M.; Bazzucchi, M.; Puggioni, G.; Brajon, G.; Dei Giudici, S.; Taccori, F.; Feliziani, F.; De Mia, G.M. Phylogenetic analysis of small ruminant lentivirus (SRLV) in Italian flocks reveals the existence of novel genetic subtypes. Virus Genes 2011, 43, 380–384. [Google Scholar] [CrossRef]
- Grego, E.; Bertolotti, L.; Quasso, A.; Profiti, M.; Lacerenza, D.; Muz, D.; Rosati, S. Genetic characterization of small ruminant lentivirus in Italian mixed flocks: Evidence for a novel genotype circulating in a local goat population. J. Gen. Virol. 2007, 88, 3423–3427. [Google Scholar] [CrossRef]
- Reina, R.; Bertolotti, L.; Dei Giudici, S.; Puggioni, G.; Ponti, N.; Profiti, M.; Patta, C.; Rosati, S. Small ruminant lentivirus genotype E is widespread in Sarda goat. Vet. Microbiol. 2010, 144, 24–31. [Google Scholar] [CrossRef] [PubMed]
- Nardelli, S.; Bettini, A.; Capello, K.; Bertoni, G.; Tavella, A. Eradication of caprine arthritis encephalitis virus in the goat population of South Tyrol, Italy: Analysis of the tailing phenomenon during the 2016–2017 campaign. J. Vet. Diagn. Investig. 2020, 32, 589–593. [Google Scholar] [CrossRef]
- Cirone, F.; Maggiolino, A.; Cirilli, M.; Sposato, A.; De Palo, P.; Ciappetta, G.; Pratelli, A. Small ruminant lentiviruses in goats in southern Italy: Serological evidence, risk factors and implementation of control programs. Vet. Microbiol. 2019, 228, 143–146. [Google Scholar] [CrossRef] [PubMed]
- Arcangeli, C.; Lucarelli, D.; Torricelli, M.; Sebastiani, C.; Ciullo, M.; Pellegrini, C.; Felici, A.; Costarelli, S.; Giammarioli, M.; Feliziani, F.; et al. First Survey of SNPs in TMEM154, TLR9, MYD88 and CCR5 Genes in Sheep Reared in Italy and Their Association with Resistance to SRLVs Infection. Viruses 2021, 13, 1290. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
- Kimura, M. A Simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian Phylogenetic Inference and Model Choice Across a Large Model Space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.-M.; Brenner, S.E. WebLogo: A Sequence Logo Generator: Figure 1. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Schneider, T.D.; Stephens, R.M. Sequence logos: A new way to display consensus sequences. Nucl. Acids Res. 1990, 18, 6097–6100. [Google Scholar] [CrossRef] [PubMed]
- Kryazhimskiy, S.; Plotkin, J.B. The Population Genetics of dN/dS. PLoS Genet. 2008, 4, e1000304. [Google Scholar] [CrossRef] [PubMed]
- Korber, B. HIV Signature and Sequence Variation Analysis. In Computational Analysis of HIV Molecular Sequences; Korber, B., Rodrigo, A.G., Learn, G.H., Eds.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2000; Chapter 4; pp. 55–72. [Google Scholar]
- Echeverría, I.; De Miguel, R.; De Pablo-Maiso, L.; Glaria, I.; Benito, A.A.; De Blas, I.; De Andrés, D.; Luján, L.; Reina, R. Multi-Platform Detection of Small Ruminant Lentivirus Antibodies and Provirus as Biomarkers of Production Losses. Front. Vet. Sci. 2020, 7, 182. [Google Scholar] [CrossRef]
- Kampen, A.H.; Åkerstedt, J.; Klevar, S. The Surveillance Programme for Small Ruminant Lentivirus Infections in Sheep and Goats in Norway 2020; Norwegian Veterinary Institute: Oslo, Norway, 2021; ISSN 1890-3290. [Google Scholar]
- De Martin, E.; Golomingi, A.; Zahno, M.; Cachim, J.; Di Labio, E.; Perler, L.; Abril, C.; Zanoni, R.; Bertoni, G. Diagnostic response to a cross-border challenge for the Swiss caprine arthritis encephalitis virus eradication program. Schweiz. Arch. Tierheilkd. 2019, 16, 93–104. [Google Scholar] [CrossRef] [PubMed]
- Adjadj, N.R.; Vicca, J.; Michiels, R.; De Regge, N. (Non-)Sense of Milk Testing in Small Ruminant Lentivirus Control Programs in Goats. Comparative Analysis of Antibody Detection and Molecular Diagnosis in Blood and Milk. Viruses 2019, 12, 3. [Google Scholar] [CrossRef] [PubMed]
- Peterhans, E.; Greenland, T.; Badiola, J.; Harkiss, G.; Bertoni, G.; Amorena, B.; Eliaszewicz, M.; Juste, R.A.; Kraßnig, R.; Lafont, J.-P.; et al. Routes of Transmission and Consequences of Small Ruminant Lentiviruses (SRLVs) Infection and Eradication Schemes. Vet. Res. 2004, 35, 257–274. [Google Scholar] [CrossRef]
- Tavella, A.; Capello, K.; Bertoni, G.; Bettini, A. Risk Factors Associated with the Alpine Multispecies Farming System in the Eradication of CAEV in South Tyrol, Italy. Viruses 2021, 13, 1959. [Google Scholar] [CrossRef]
- Cvijović, I.; Good, B.H.; Desai, M.M. The Effect of Strong Purifying Selection on Genetic Diversity. Genetics 2018, 209, 1235–1278. [Google Scholar] [CrossRef]
- Rosati, S.; Mannelli, A.; Merlo, T.; Ponti, N. Characterization of the immunodominant cross-reacting epitope of visna maedi virus and caprine arthritis-encephalitis virus capsid antigen. Virus Res. 1999, 61, 177–183. [Google Scholar] [CrossRef]
- Tanaka, M.; Robinson, B.A.; Chutiraka, K.; Geary, C.D.; Reed, J.C.; Lingappa, J.R. Mutations of Conserved Residues in the Major Homology Region Arrest Assembling HIV-1 Gag as a Membrane-Targeted Intermediate Containing Genomic RNA and Cellular Proteins. J. Virol. 2016, 90, 1944–1963. [Google Scholar] [CrossRef]
- Chu, H.-H.; Chang, Y.-F.; Wang, C.-T. Mutations in the α-helix Directly C-terminal to the Major Homology Region of Human Immunodeficiency Virus Type 1 Capsid Protein Disrupt Gag Multimerization and Markedly Impair Virus Particle Production. J. Biomed. Sci. 2006, 13, 645–656. [Google Scholar] [CrossRef] [PubMed]
- Minardi da Cruz, J.; Singh, D.; Lamara, A.; Chebloune, Y. Small Ruminant Lentiviruses (SRLVs) Break the Species Barrier to Acquire New Host Range. Viruses 2013, 5, 1867–1884. [Google Scholar] [CrossRef] [PubMed]
- Barros, S.C.; Ramos, F.; Duarte, M.; Fagulha, T.; Cruz, B.; Fevereiro, M. Genomic Characterization of a Slow/Low Maedi Visna Virus. Virus Genes 2004, 29, 199–210. [Google Scholar] [CrossRef] [PubMed]
- Staskus, K.A.; Retzel, E.F.; Lewis, E.D.; Silsby, J.L.; Cyr, S.S.; Rank, J.M.; Wietgrefe, S.W.; Haase, A.T.; Cook, R.; Fast, D.; et al. Isolation of replication-competent molecular clones of visna virus. Virology 1991, 181, 228–240. [Google Scholar] [CrossRef]
- Braun, M.J.; Clements, J.E.; Gonda, M.A. The visna virus genome: Evidence for a hypervariable site in the env gene and sequence homology among lentivirus envelope proteins. J. Virol. 1987, 61, 4046–4054. [Google Scholar] [CrossRef]
- Grego, E.; Bertolotti, L.; Carrozza, M.; Profiti, M.; Mazzei, M.; Tolari, F.; Rosati, S. Genetic and antigenic characterization of the matrix protein of two genetically distinct ovine lentiviruses. Veter. Microbiol. 2005, 106, 179–185. [Google Scholar] [CrossRef]
- Herrmann-Hoesing, L.M.; Broughton-Neiswanger, L.E.; Gouine, K.C.; White, S.N.; Mousel, M.R.; Lewis, G.S.; Marshall, K.L.; Knowles, D.P. Evaluation of a Caprine Arthritis-Encephalitis Virus/Maedi-Visna Virus Indirect Enzyme-Linked Immunosorbent Assay in the Serological Diagnosis of Ovine Progressive Pneumonia Virus in U.S. Sheep. Clin. Vaccine Immunol. 2010, 17, 307–310. [Google Scholar] [CrossRef]
- Huang, J.; Sun, Y.; Liu, Y.; Xiao, H.; Zhuang, S. Development of a loop-mediated isothermal amplification method for rapid detection of caprine arthritis-encephalitis virus proviral DNA. 2012, 157, 1463–1469. Arch. Virol. 2012, 157, 1463–1469. [Google Scholar] [CrossRef]
- Saltarelli, M.; Querat, G.; Konings, D.A.; Vigne, R.; Clements, J.E. Nucleotide sequence and transcriptional analysis of molecular clones of CAEV which generate infectious virus. Virology 1990, 179, 347–364. [Google Scholar] [CrossRef]
- Ramírez, H.; Glaria, I.; de Andrés, X.; Martínez, H.; Hernández, M.; Reina, R.; Iráizoz, E.; Crespo, H.; Berriatua, E.; Vázquez, J.; et al. Recombinant small ruminant lentivirus subtype B1 in goats and sheep of imported breeds in Mexico. Vet. J. 2011, 190, 169–172. [Google Scholar] [CrossRef]
- Glaria, I.; Reina, R.; Crespo, H.; de Andrés, X.; Ramírez, H.; Biescas, E.; Pérez, M.M.; Badiola, J.; Luján, L.; Amorena, B.; et al. Phylogenetic analysis of SRLV sequences from an arthritic sheep outbreak demonstrates the introduction of CAEV-like viruses among Spanish sheep. Vet. Microbiol. 2009, 138, 156–162. [Google Scholar] [CrossRef] [PubMed]
- Reina, R.; Grego, E.; Bertolotti, L.; De Meneghi, D.; Rosati, S. Genome Analysis of Small-Ruminant Lentivirus Genotype E: A Caprine Lentivirus with Natural Deletions of the dUTPase Subunit, vpr -Like Accessory Gene, and 70-Base-Pair Repeat of the U3 Region. J. Virol. 2009, 83, 1152–1155. [Google Scholar] [CrossRef] [PubMed]
- Glaria, I.; Reina, R.; Ramírez, H.; de Andrés, X.; Crespo, H.; Jáuregui, P.; Salazar, E.; Luján, L.; Pérez, M.; Benavides, J.; et al. Visna/Maedi virus genetic characterization and serological diagnosis of infection in sheep from a neurological outbreak. Vet. Microbiol. 2012, 155, 137–146. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arcangeli, C.; Torricelli, M.; Sebastiani, C.; Lucarelli, D.; Ciullo, M.; Passamonti, F.; Giammarioli, M.; Biagetti, M. Genetic Characterization of Small Ruminant Lentiviruses (SRLVs) Circulating in Naturally Infected Sheep in Central Italy. Viruses 2022, 14, 686. https://doi.org/10.3390/v14040686
Arcangeli C, Torricelli M, Sebastiani C, Lucarelli D, Ciullo M, Passamonti F, Giammarioli M, Biagetti M. Genetic Characterization of Small Ruminant Lentiviruses (SRLVs) Circulating in Naturally Infected Sheep in Central Italy. Viruses. 2022; 14(4):686. https://doi.org/10.3390/v14040686
Chicago/Turabian StyleArcangeli, Chiara, Martina Torricelli, Carla Sebastiani, Daniele Lucarelli, Marcella Ciullo, Fabrizio Passamonti, Monica Giammarioli, and Massimo Biagetti. 2022. "Genetic Characterization of Small Ruminant Lentiviruses (SRLVs) Circulating in Naturally Infected Sheep in Central Italy" Viruses 14, no. 4: 686. https://doi.org/10.3390/v14040686
APA StyleArcangeli, C., Torricelli, M., Sebastiani, C., Lucarelli, D., Ciullo, M., Passamonti, F., Giammarioli, M., & Biagetti, M. (2022). Genetic Characterization of Small Ruminant Lentiviruses (SRLVs) Circulating in Naturally Infected Sheep in Central Italy. Viruses, 14(4), 686. https://doi.org/10.3390/v14040686