
Effective Reduction of SARS-CoV-2 RNA Levels Using a Tailor-Made Oligonucleotide-Based RNA Inhibitor

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Oligonucleotides
2.2. Hybridization
2.3. Vector Construction
2.4. Bacterial Transformation and Vector Cloning
2.5. Cell Culture
2.6. Transfection of Cells
2.7. Treatment of Cells
2.8. Confocal Microscopy
2.9. Quantification of Cellular Uptake
2.10. Collection of Patient Samples
2.11. Treatment of Patient Samples
2.12. RNA Isolation
2.13. Real-Time RT-PCR
3. Results and Discussion
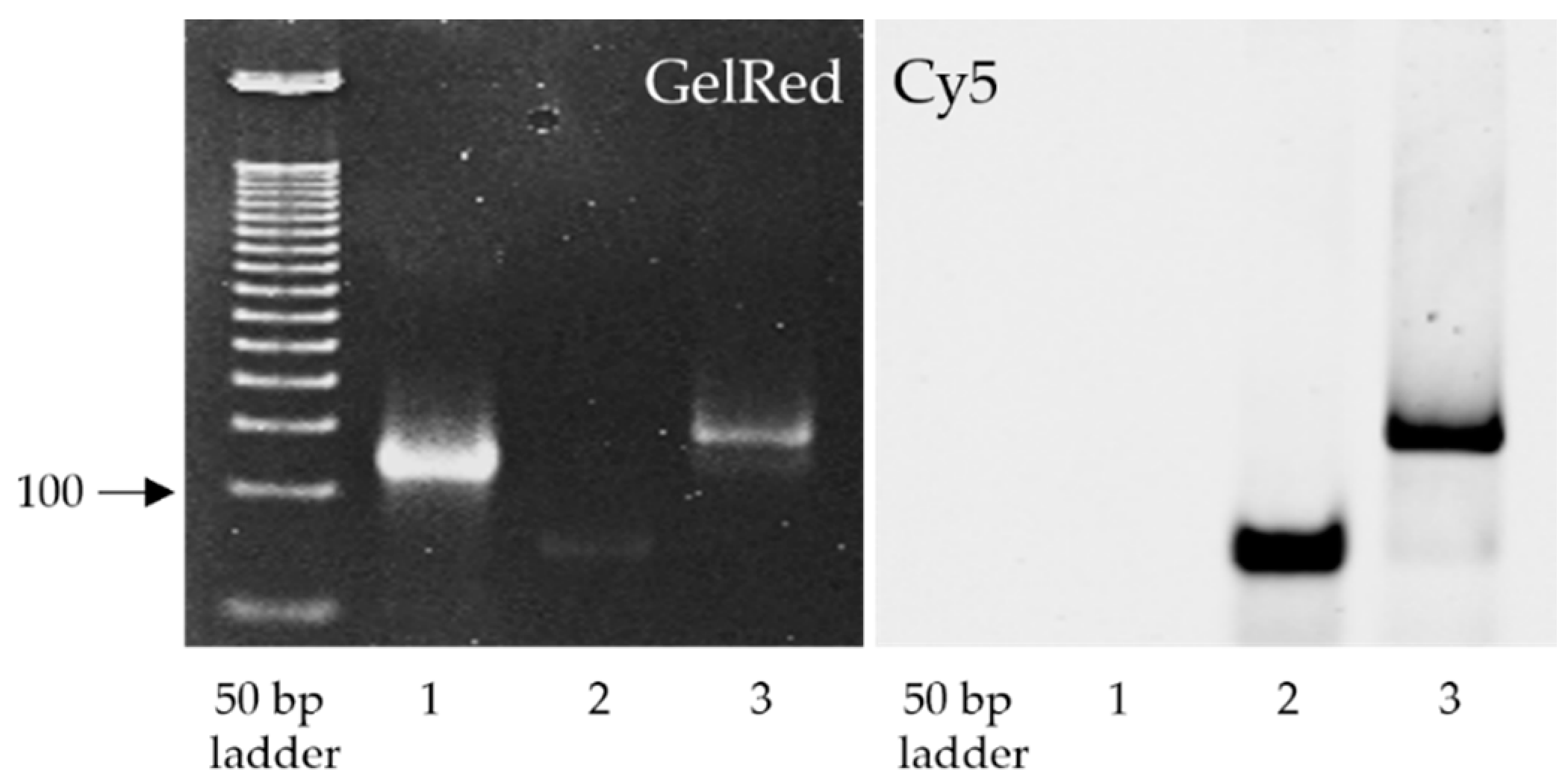
3.1. Binding of ASC1R to the Target SARS-CoV-2 RNA Region
3.2. Cellular Internalization of ASC1R
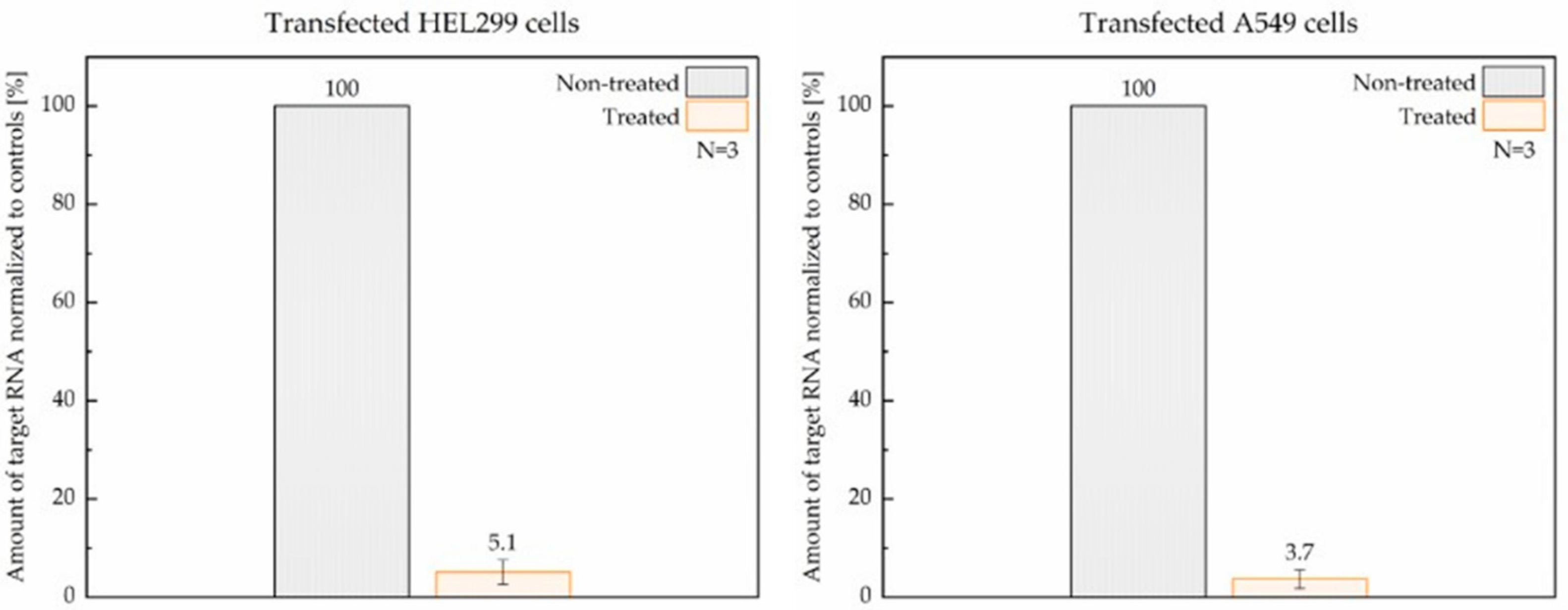
3.3. Efficacy of ASC1R in Transfected HEL299 and A549 Cells
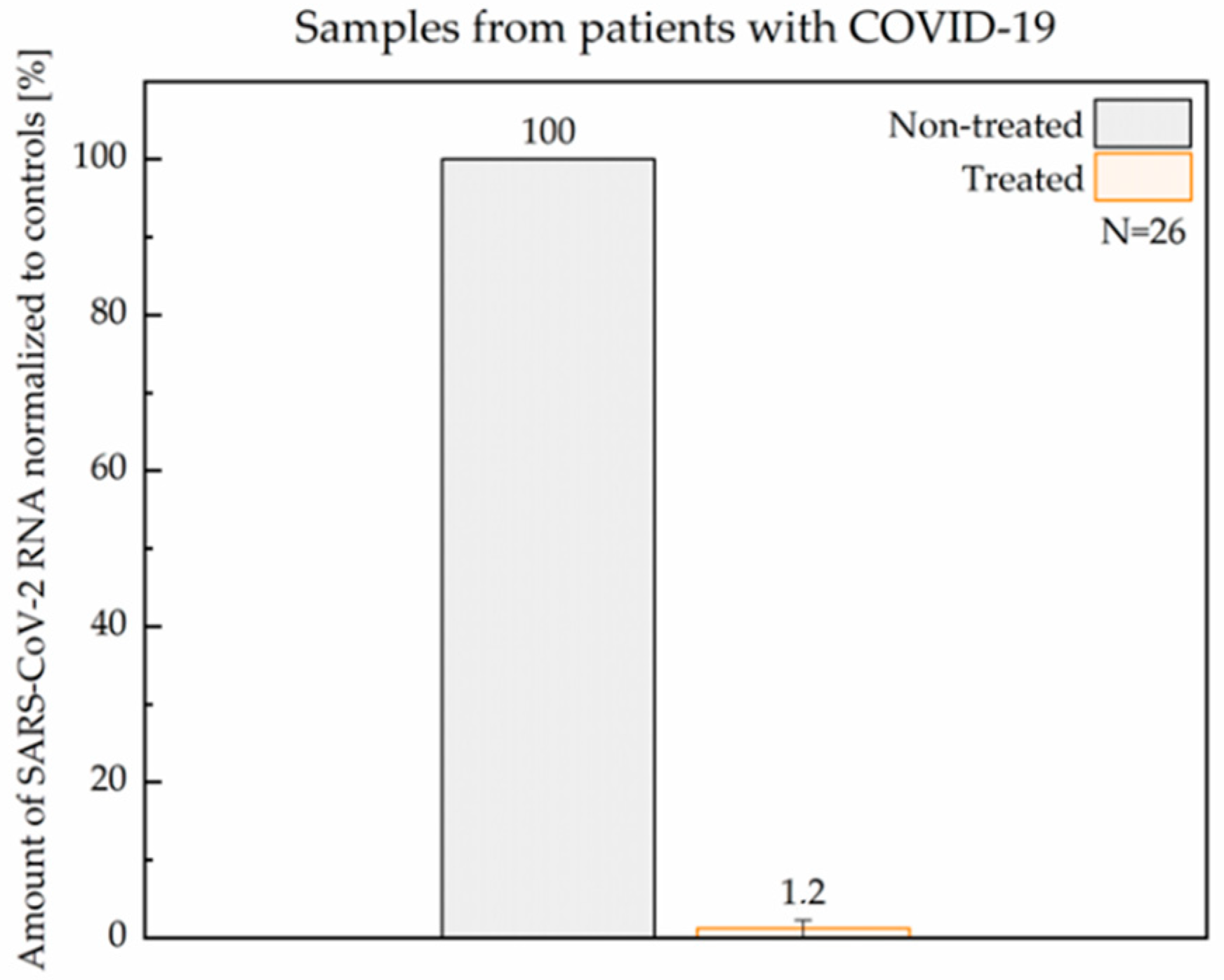
3.4. Efficacy of ASC1R in Patient Samples
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- WHO Coronavirus (COVID-19) Dashboard. Available online: https://covid19.who.int/ (accessed on 10 February 2022).
- Talic, S.; Shah, S.; Wild, H.; Gasevic, D.; Maharaj, A.; Ademi, Z.; Li, X.; Xu, W.; Mesa-Eguiagaray, I.; Rostron, J.; et al. Effectiveness of public health measures in reducing the incidence of covid-19, SARS-CoV-2 transmission, and covid-19 mortality: Systematic review and meta-analysis. Br. Med. J. 2021, 375, e068302. [Google Scholar]
- Tregoning, J.S.; Flight, K.E.; Higham, S.L.; Wang, Z.; Pierce, B.F. Progress of the COVID-19 vaccine effort: Viruses, vaccines and variants versus efficacy, effectiveness and escape. Nat. Rev. Immunol. 2021, 21, 626–636. [Google Scholar] [CrossRef] [PubMed]
- Kyriakidis, N.C.; López-Cortés, A.; González, E.V.; Grimaldos, A.B.; Prado, E.O. SARS-CoV-2 vaccines strategies: A comprehensive review of phase 3 candidates. NPJ Vaccines 2021, 6, 28. [Google Scholar] [CrossRef] [PubMed]
- Cully, M. A tale of two antiviral targets—And the COVID-19 drugs that bind them. Nat. Rev. Drug. Discov. 2022, 21, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Kröker, A.; Tirzīte, M. Repurposed pharmacological agents for the potential treatment of COVID-19: A literature review. Respir. Res. 2021, 22, 304. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.; Tang, Z.; Zhao, J.; Wang, J. Inhibition of SARS-CoV-2 by Targeting Conserved Viral RNA Structures and Sequences. Front. Chem. 2021, 9, 802766. [Google Scholar] [CrossRef] [PubMed]
- Aftab, S.O.; Ghouri, M.Z.; Masood, M.U.; Haider, Z.; Khan, Z.; Ahmad, A.; Munawar, N. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J. Transl. Med. 2020, 18, 275. [Google Scholar] [CrossRef] [PubMed]
- Quemener, A.M.; Centomo, M.L.; Sax, S.L.; Panella, R. Small drugs, huge impact: The extraordinary impact of antisense oligonucleotides in research and drug development. Molecules 2022, 27, 536. [Google Scholar] [CrossRef] [PubMed]
- Karaki, S.; Paris, C.; Rocchi, P. Antisense oligonucleotides, a novel developing targeting therapy. In Antisense Therapy; Sharad, S., Ed.; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Rinaldi, C.; Wood, M.J.A. Antisense oligonucleotides: The next frontier for treatment of neurological disorders. Nat. Rev. Neurol. 2018, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Razga, F.; Nemethova, V. Selective therapeutic intervention: A challenge against off-target effects. Trends Mol. Med. 2017, 23, 671–674. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.C.; Goddard, E.T.; Lanfranca, M.P.; Davido, D.J. hTERT extends the life of human fibroblasts without compromising type I interferon signaling. PLoS ONE 2013, 8, e58233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smetsers, T.F.; Skorski, T.; van de Locht, L.T.; Wessels, H.M.; Pennings, A.H.; de Witte, T.; Calabretta, B.; Mensing, E.J. Antisense BCR-ABL oligonucleotides induce apoptosis in the Philadelphia chromosome-positive cell line BV173. Leukemia 1994, 8, 129–140. [Google Scholar] [PubMed]
- Kapustin, A.N.; Davey, P.; Longmire, D.; Matthews, C.; Linnane, E.; Rustogi, N.; Stavrou, M.; Devine, P.W.A.; Bond, N.J.; Hanson, L.; et al. Antisense oligonucleotide activity in tumour cells is influenced by intracellular LBPA distribution and extracellular vesicle recycling. Commun. Biol. 2021, 4, 1241. [Google Scholar] [CrossRef] [PubMed]
- Koller, E.; Vincent, T.M.; Chappell, A.; De, S.; Manoharan, M.; Bennett, C.F. Mechanisms of single-stranded phosphorothioate modified antisense oligonucleotide accumulation in hepatocytes. Nucleic Acids Res. 2011, 39, 4795–4807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pendergraff, H.; Schmidt, S.; Vikesa, J.; Weile, C.; Øverup, C.; Lindholm, M.W.; Koch, T. Nuclear and cytoplasmatic quantification of unconjugated, label-free locked nucleic acid oligonucleotides. Nucleic Acid Ther. 2020, 30, 4–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, X.H.; Sun, H.; Nichols, J.G.; Crooke, S.T. RNase H1-dependent antisense oligonucleotides are robustly active in directing RNA cleavage in both the cytoplasm and the nucleus. Mol. Ther. 2017, 25, 2075–2092. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nemethova, V.; Mazancova, P.; Selc, M.; Jakic, K.; Uhelska, L.; Nemethova, B.; Poturnayova, A.; Drgona, L.; Babelova, A.; Razga, F. Effective Reduction of SARS-CoV-2 RNA Levels Using a Tailor-Made Oligonucleotide-Based RNA Inhibitor. Viruses 2022, 14, 685. https://doi.org/10.3390/v14040685
Nemethova V, Mazancova P, Selc M, Jakic K, Uhelska L, Nemethova B, Poturnayova A, Drgona L, Babelova A, Razga F. Effective Reduction of SARS-CoV-2 RNA Levels Using a Tailor-Made Oligonucleotide-Based RNA Inhibitor. Viruses. 2022; 14(4):685. https://doi.org/10.3390/v14040685
Chicago/Turabian StyleNemethova, Veronika, Petra Mazancova, Michal Selc, Kristina Jakic, Lucia Uhelska, Boglarka Nemethova, Alexandra Poturnayova, Lubos Drgona, Andrea Babelova, and Filip Razga. 2022. "Effective Reduction of SARS-CoV-2 RNA Levels Using a Tailor-Made Oligonucleotide-Based RNA Inhibitor" Viruses 14, no. 4: 685. https://doi.org/10.3390/v14040685
APA StyleNemethova, V., Mazancova, P., Selc, M., Jakic, K., Uhelska, L., Nemethova, B., Poturnayova, A., Drgona, L., Babelova, A., & Razga, F. (2022). Effective Reduction of SARS-CoV-2 RNA Levels Using a Tailor-Made Oligonucleotide-Based RNA Inhibitor. Viruses, 14(4), 685. https://doi.org/10.3390/v14040685