
Presence of Recombinant Bat Coronavirus GCCDC1 in Cambodian Bats

 , , , , , , , , and
, , , , , , , , and 
Abstract
:1. Introduction
2. Materials and Methods
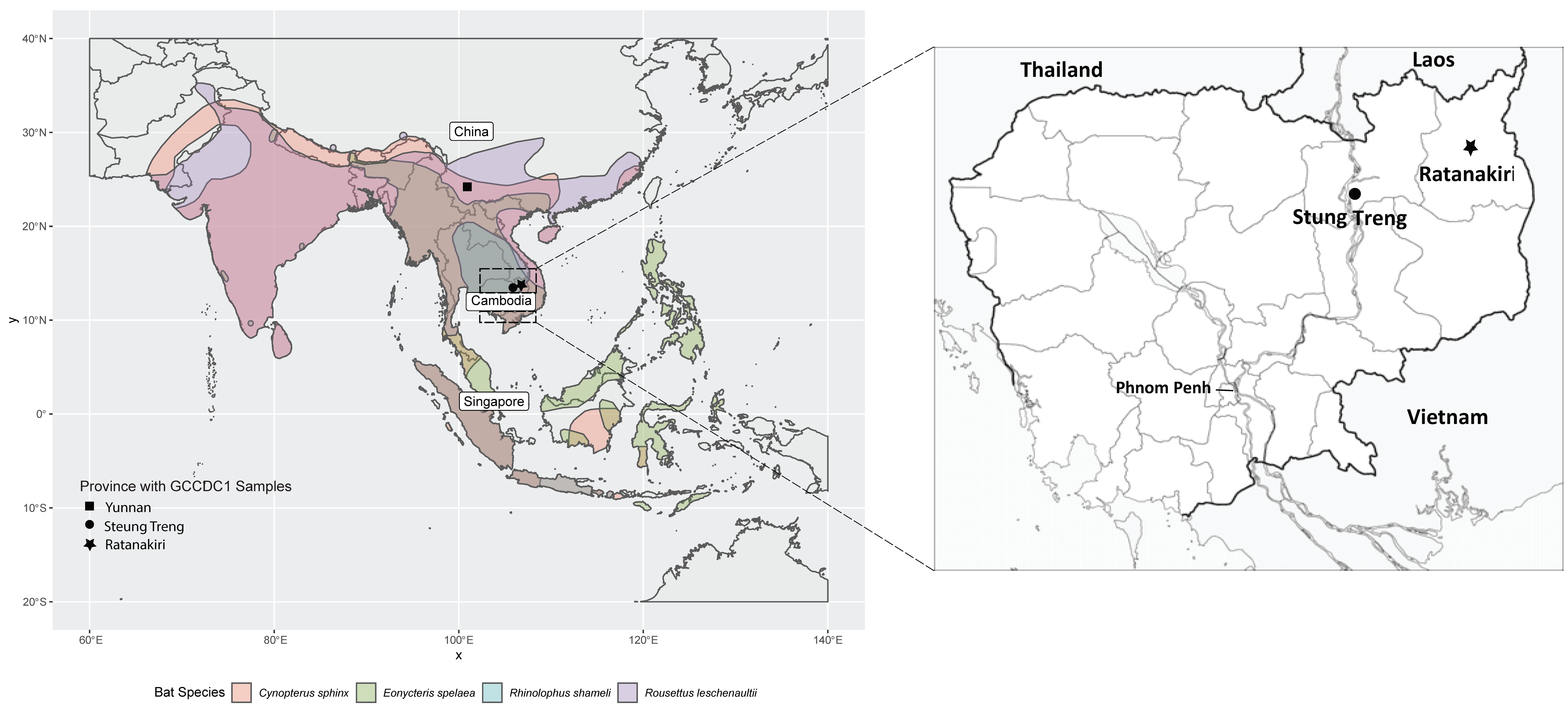
2.1. Sample Collection
2.2. RNA Extraction
2.3. Preparation of Illumina DNA Libraries from RNA
2.4. Virus Enrichment
2.5. Data Analysis
3. Results
3.1. Sample Collection, Virus Isolation, RNA Extraction and NGS Analysis
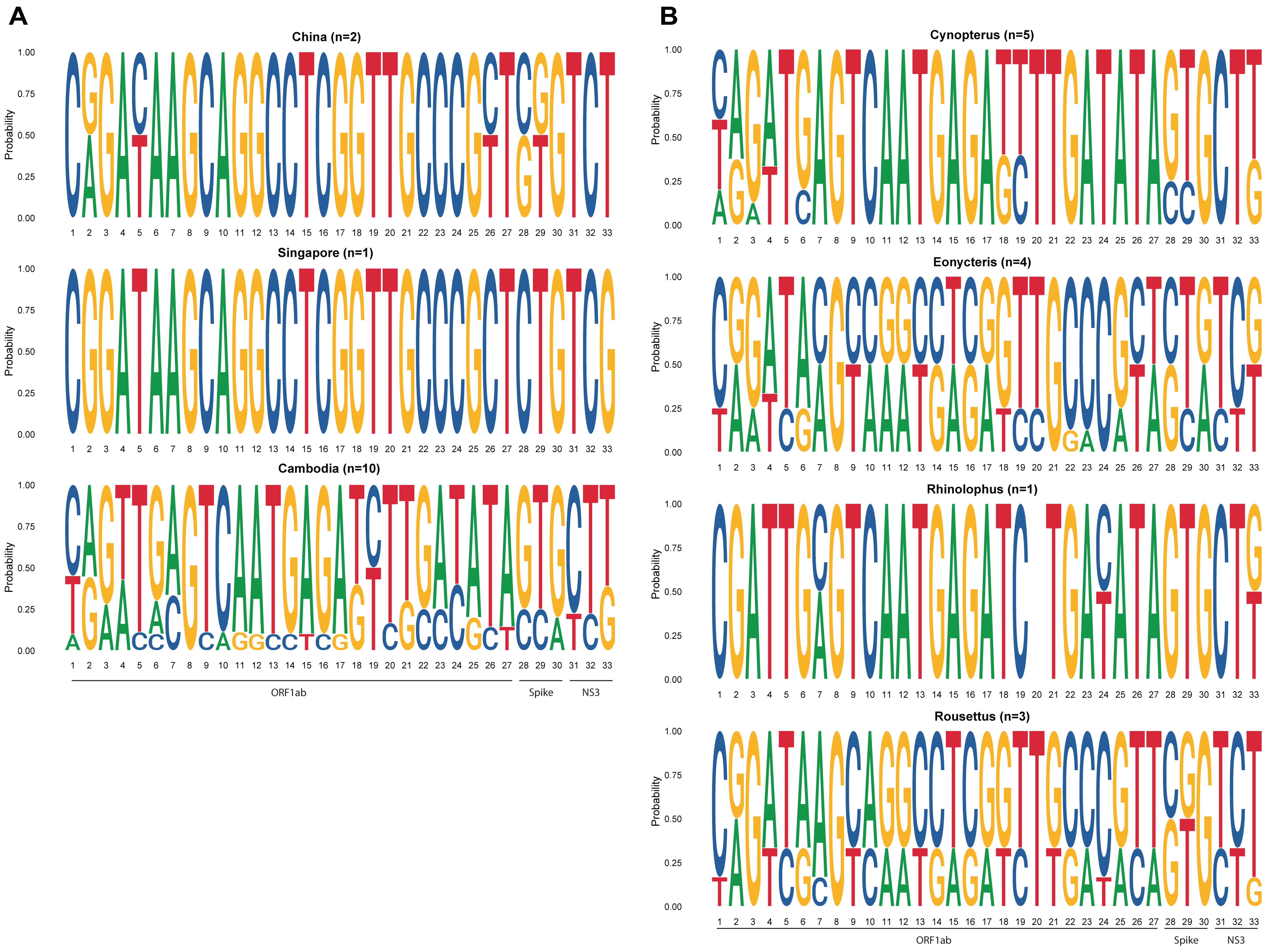
3.2. Bat Species-Specific Features
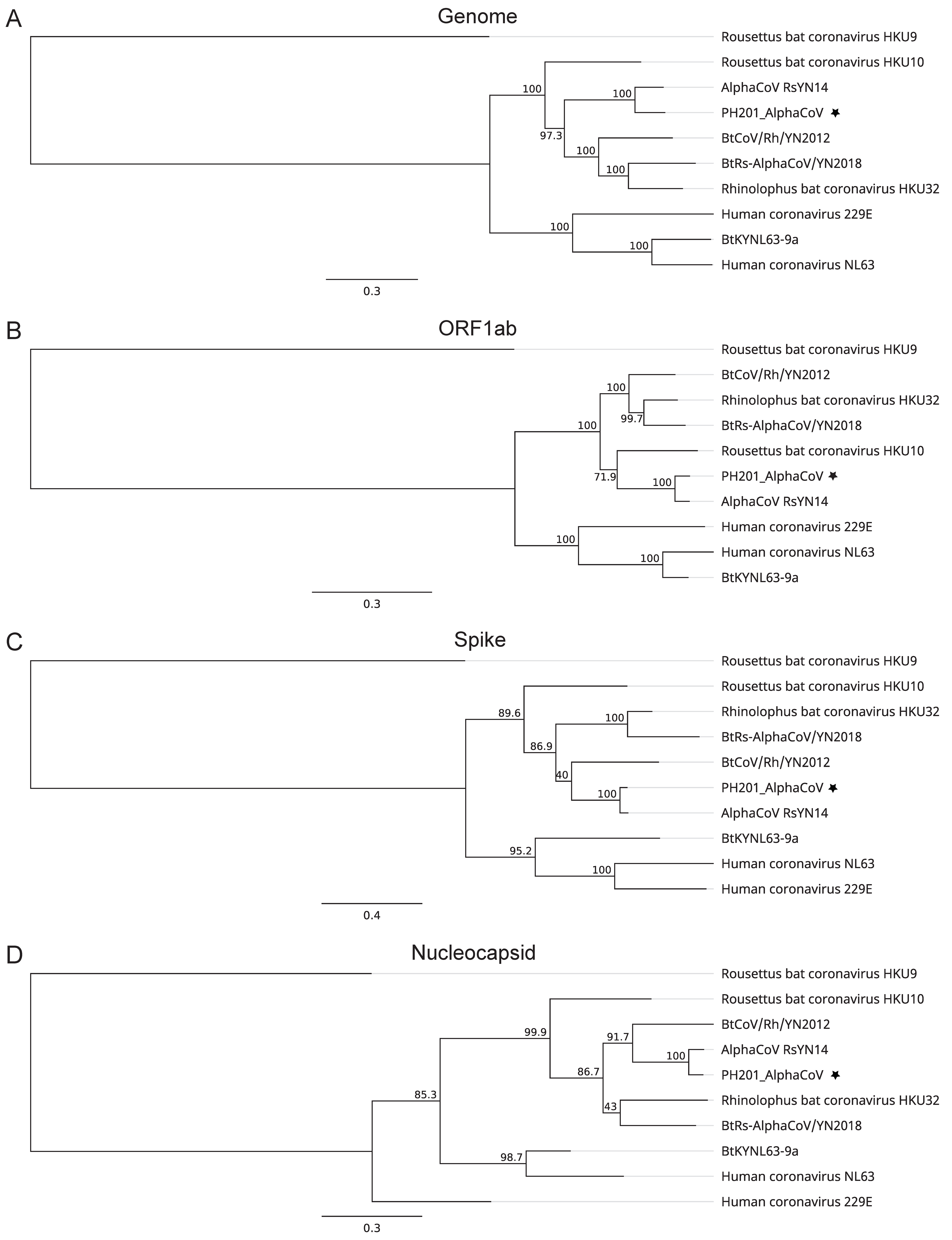
3.3. RsYN14-Like AlphaCoV in PH201
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Rota, P.A.; Oberste, M.S.; Monroe, S.S.; Nix, W.A.; Campagnoli, R.; Icenogle, J.P.; Penaranda, S.; Bankamp, B.; Maher, K.; Chen, M.H.; et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 2003, 300, 1394–1399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Haagmans, B.L.; Al Dhahiry, S.H.; Reusken, C.B.; Raj, V.S.; Galiano, M.; Myers, R.; Godeke, G.J.; Jonges, M.; Farag, E.; Diab, A.; et al. Middle East respiratory syndrome coronavirus in dromedary camels: An outbreak investigation. Lancet Infect. Dis. 2014, 14, 140–145. [Google Scholar] [CrossRef] [Green Version]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Guan, X.; Wu, P.; Wang, X.; Zhou, L.; Tong, Y.; Ren, R.; Leung, K.S.M.; Lau, E.H.Y.; Wong, J.Y.; et al. Early Transmission Dynamics in Wuhan, China, of Novel Coronavirus-Infected Pneumonia. N. Engl. J. Med. 2020, 382, 1199–1207. [Google Scholar] [CrossRef] [PubMed]
- Worldometer. COVID-19 Coronavirus Pandemic. Available online: https://www.worldometers.info/coronavirus/ (accessed on 31 December 2021).
- Walker, P.J.; Siddell, S.G.; Lefkowitz, E.J.; Mushegian, A.R.; Adriaenssens, E.M.; Alfenas-Zerbini, P.; Davison, A.J.; Dempsey, D.M.; Dutilh, B.E.; García, M.L.; et al. Changes to virus taxonomy and to the International Code of Virus Classification and Nomenclature ratified by the International Committee on Taxonomy of Viruses (2021). Arch. Virol. 2021, 166, 2633–2648. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef] [PubMed]
- Bobay, L.-M.; O’Donnell, A.C.; Ochman, H. Recombination events are concentrated in the spike protein region of Betacoronaviruses. PLoS Genet. 2020, 16, e1009272. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Liu, W.J.; Xu, W.; Jin, T.; Zhao, Y.; Song, J.; Shi, Y.; Ji, W.; Jia, H.; Zhou, Y.; et al. A Bat-Derived Putative Cross-Family Recombinant Coronavirus with a Reovirus Gene. PLoS Pathog. 2016, 12, e1005883. [Google Scholar] [CrossRef] [PubMed]
- Paskey, A.C.; Ng, J.H.J.; Rice, G.K.; Chia, W.N.; Philipson, C.W.; Foo, R.J.H.; Cer, R.Z.; Long, K.A.; Lueder, M.R.; Lim, X.F.; et al. Detection of Recombinant Rousettus Bat Coronavirus GCCDC1 in Lesser Dawn Bats (Eonycteris spelaea) in Singapore. Viruses 2020, 12, 539. [Google Scholar] [CrossRef]
- Lim, X.F.; Lee, C.B.; Pascoe, S.M.; How, C.B.; Chan, S.; Tan, J.H.; Yang, X.; Zhou, P.; Shi, Z.; Sessions, O.M.; et al. Detection and characterization of a novel bat-borne coronavirus in Singapore using multiple molecular approaches. J. Gen. Virol. 2019, 100, 1363–1374. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Ji, J.; Chen, X.; Bi, Y.; Li, J.; Wang, Q.; Hu, T.; Song, H.; Zhao, R.; Chen, Y.; et al. Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 2021, 184, 4380–4391.e14. [Google Scholar] [CrossRef] [PubMed]
- Delaune, D.; Hul, V.; Karlsson, E.A.; Hassanin, A.; Ou, T.P.; Baidaliuk, A.; Gambaro, F.; Prot, M.; Tu, V.T.; Chea, S.; et al. A novel SARS-CoV-2 related coronavirus in bats from Cambodia. Nat. Commun. 2021, 12, 6563. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Animal ID | Country | Province | Village | Family | Genus | Species | Type of Sample | Date Sample Taken |
|---|---|---|---|---|---|---|---|---|
| PH162 | Cambodia | Steung Treng | Chhab Village | Pteropodidae | Cynopterus | sp. | Rectal swab | 5–6-December-2010 |
| PH178 | Cambodia | Steung Treng | Chhab Village | Pteropodidae | Rousettus | sp. | Oral swab | 6–7-December-2010 |
| PH201 | Cambodia | Steung Treng | Chhab Village | Rhinolophidae | Rhinolophus | shameli | Rectal swab | 6–7-December-2010 |
| RK022 | Cambodia | Ratanakiri | Pouy Village | Pteropodidae | Cynopterus | sp. | Rectal swab | 14–15-December-2010 |
| RK058 | Cambodia | Ratanakiri | Ta Kouy Village | Pteropodidae | Eonycteris | spelaea | Rectal swab | 15–16-December-2010 |
| RK059 | Cambodia | Ratanakiri | Ta Kouy Village | Pteropodidae | Cynopterus | sphinx | Rectal swab | 15–16-December-2010 |
| RK072 | Cambodia | Ratanakiri | Veung Seing Village | Pteropodidae | Cynopterus | sp. | Rectal swab | 17–18-December-2010 |
| RK074 | Cambodia | Ratanakiri | Veung Seing Village | Pteropodidae | Eonycteris | spelaea | Rectal swab | 17–18-December-2010 |
| RK075 | Cambodia | Ratanakiri | Pan Porng Village | Pteropodidae | Cynopterus | sp. | Rectal swab | 18–19-December-2010 |
| RK091 | Cambodia | Ratanakiri | Pan Porng Village | Pteropodidae | Eonycteris | spelaea | Rectal swab | 18–19-December-2010 |
| MT350598 (GCCDC1 Singapore) | NC_030886 (GCCDC1 356) | KU762337 (GCCDC1 346) | ORF1ab | S | NS3 | E | M | N | p10 | NS7a | NS7b | NS7c | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ID | Length | X# | Genome | Genome | Genome | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# |
| PH162 | 30101 | 90.9 | 90.49 | 92 | 93.81 | 90.9 | 90.5 | 92 | 93.8 | 94.8 | 97 | 97.5 | 97 | 100 | 27.9 | 8.31 | 3.9 | 86.7 | 84.8 | 87 | 96.5 | 97.7 | 100 | 98.2 | 98.9 | 100 | 97.3 | 100 | 100 | 97.9 | 98.5 | 100 | 66.3 | 57.1 | 56 |
| PH178 | 30101 | 93 | 92.84 | 94.4 | 92.07 | 93 | 92.8 | 94 | 92.1 | 93.1 | 94 | 97.5 | 97.4 | 100 | 28.9 | 8.31 | 5.2 | 97.4 | 97.7 | 100 | 96.5 | 97.7 | 100 | 98.2 | 98.9 | 100 | 97.3 | 100 | 100 | 97.9 | 98.5 | 100 | 73.6 | 60.1 | 69 |
| PH201 | 30134 | 94.2 | 94.8 | 95.6 | 93.53 | 94.2 | 94.8 | 96 | 93.5 | 95.2 | 96 | 97.6 | 97.4 | 100 | 69.2 | 47.3 | 75 | 97.3 | 97.7 | 100 | 96.5 | 97.7 | 100 | 98.2 | 98.9 | 100 | 97.3 | 100 | 100 | 97.9 | 98.5 | 100 | 97.3 | 96.8 | 98 |
| RK022 | 30113 | 82.8 | 77.79 | 81.8 | 83.93 | 82.8 | 77.8 | 82 | 83.9 | 81.1 | 83 | 89.4 | 85.7 | 90 | 27.9 | 8.31 | 3.9 | 82.2 | 78 | 78 | 96.4 | 97.5 | 100 | 97.5 | 96.8 | 100 | 97.3 | 100 | 100 | 97.9 | 98.5 | 100 | 78.1 | 68.9 | 75 |
| RK058 | 30139 | 97.4 | 99.12 | 100 | 96.67 | 97.4 | 99.1 | 100 | 96.7 | 99.4 | 100 | 97.8 | 98.3 | 100 | 100 | 100 | 100 | 97.4 | 97.7 | 100 | 96.5 | 97.7 | 100 | 98.2 | 98.9 | 100 | 97.3 | 100 | 100 | 97.9 | 98.5 | 100 | 86.2 | 83.3 | 82 |
| RK059 | 30101 | 93.8 | 94.31 | 95.9 | 91.01 | 93.8 | 94.3 | 96 | 91 | 91.7 | 93 | 92.8 | 91.9 | 94 | 25 | 4.55 | 0 | 92 | 90.5 | 91 | 96.5 | 97.7 | 100 | 98.2 | 98.9 | 100 | 97.3 | 100 | 100 | 97.9 | 98.5 | 100 | 97.5 | 96.1 | 98 |
| RK072 | 29961 | 63.1 | 51.22 | 55.4 | 51.64 | 63.1 | 51.2 | 55 | 51.6 | 36.7 | 41 | 53.7 | 39.6 | 42 | 25 | 4.55 | 0 | 82.6 | 70.1 | 86 | 64.4 | 52.5 | 60 | 94.8 | 89.3 | 100 | 93.8 | 93.8 | 96 | 79.8 | 87.7 | 92 | 50.3 | 35.7 | 39 |
| RK074 | 30145 | 97.6 | 99.43 | 100 | 96.45 | 97.6 | 99.4 | 100 | 96.5 | 99.2 | 100 | 97.2 | 97.4 | 100 | 100 | 100 | 100 | 97.3 | 97.7 | 100 | 96.5 | 97.7 | 100 | 98.2 | 98.9 | 100 | 97.3 | 100 | 100 | 97.9 | 98.5 | 100 | 99.6 | 100 | 100 |
| RK075 | 30147 | 94.9 | 96.2 | 97.2 | 95.29 | 94.9 | 96.2 | 97 | 95.3 | 97.4 | 98 | 97.3 | 97 | 100 | 28.9 | 8.31 | 5.2 | 97.3 | 97.7 | 100 | 96.5 | 97.7 | 100 | 98.2 | 98.9 | 100 | 97.3 | 100 | 100 | 97.9 | 98.5 | 100 | 85.4 | 82.1 | 81 |
| RK091 | 30147 | 94.3 | 94 | 96.1 | 94.04 | 94.3 | 94 | 96 | 94 | 95.1 | 98 | 96.1 | 92 | 100 | 98.3 | 100 | 100 | 96.4 | 96.8 | 100 | 98.1 | 99.6 | 100 | 98.2 | 98.9 | 100 | 97.3 | 100 | 100 | 99.5 | 98.8 | 100 | 52.3 | 39.8 | 36 |
| ID | Length | X# | MZ08395 (RsYN14) | ORF1ab | S | ORF3a | ORF4a | E | M | N | ORF8 | ORF9 | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | nt | aa | x# | ||||
| PH201 | 28807 | 96.7 | 83.23 | 82.67 | 90.37 | 96.9 | 80.36 | 91.09 | 96.9 | 85.67 | 90.61 | 100 | 77.94(early termination) | 76.29(early termination) | 100 | 90.89 | 93.55 | 93.8 | 95.22 | 98.35 | 100 | 86.75 | 91.48 | 100 | 89.89 | 90.91 | 100 | 83.77 | 80.79 | 100 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, F.; Duong, V.; Lim, X.F.; Hul, V.; Chawla, T.; Keatts, L.; Goldstein, T.; Hassanin, A.; Tu, V.T.; Buchy, P.; et al. Presence of Recombinant Bat Coronavirus GCCDC1 in Cambodian Bats. Viruses 2022, 14, 176. https://doi.org/10.3390/v14020176
Zhu F, Duong V, Lim XF, Hul V, Chawla T, Keatts L, Goldstein T, Hassanin A, Tu VT, Buchy P, et al. Presence of Recombinant Bat Coronavirus GCCDC1 in Cambodian Bats. Viruses. 2022; 14(2):176. https://doi.org/10.3390/v14020176
Chicago/Turabian StyleZhu, Feng, Veasna Duong, Xiao Fang Lim, Vibol Hul, Tanu Chawla, Lucy Keatts, Tracey Goldstein, Alexandre Hassanin, Vuong Tan Tu, Philippe Buchy, and et al. 2022. "Presence of Recombinant Bat Coronavirus GCCDC1 in Cambodian Bats" Viruses 14, no. 2: 176. https://doi.org/10.3390/v14020176
APA StyleZhu, F., Duong, V., Lim, X. F., Hul, V., Chawla, T., Keatts, L., Goldstein, T., Hassanin, A., Tu, V. T., Buchy, P., Sessions, O. M., Wang, L.-F., Dussart, P., & Anderson, D. E. (2022). Presence of Recombinant Bat Coronavirus GCCDC1 in Cambodian Bats. Viruses, 14(2), 176. https://doi.org/10.3390/v14020176