



Kiwira Virus, a Newfound Hantavirus Discovered in Free-tailed Bats (Molossidae) in East and Central Africa
 , , ,
, , ,
Abstract
1. Introduction
2. Materials and Methods
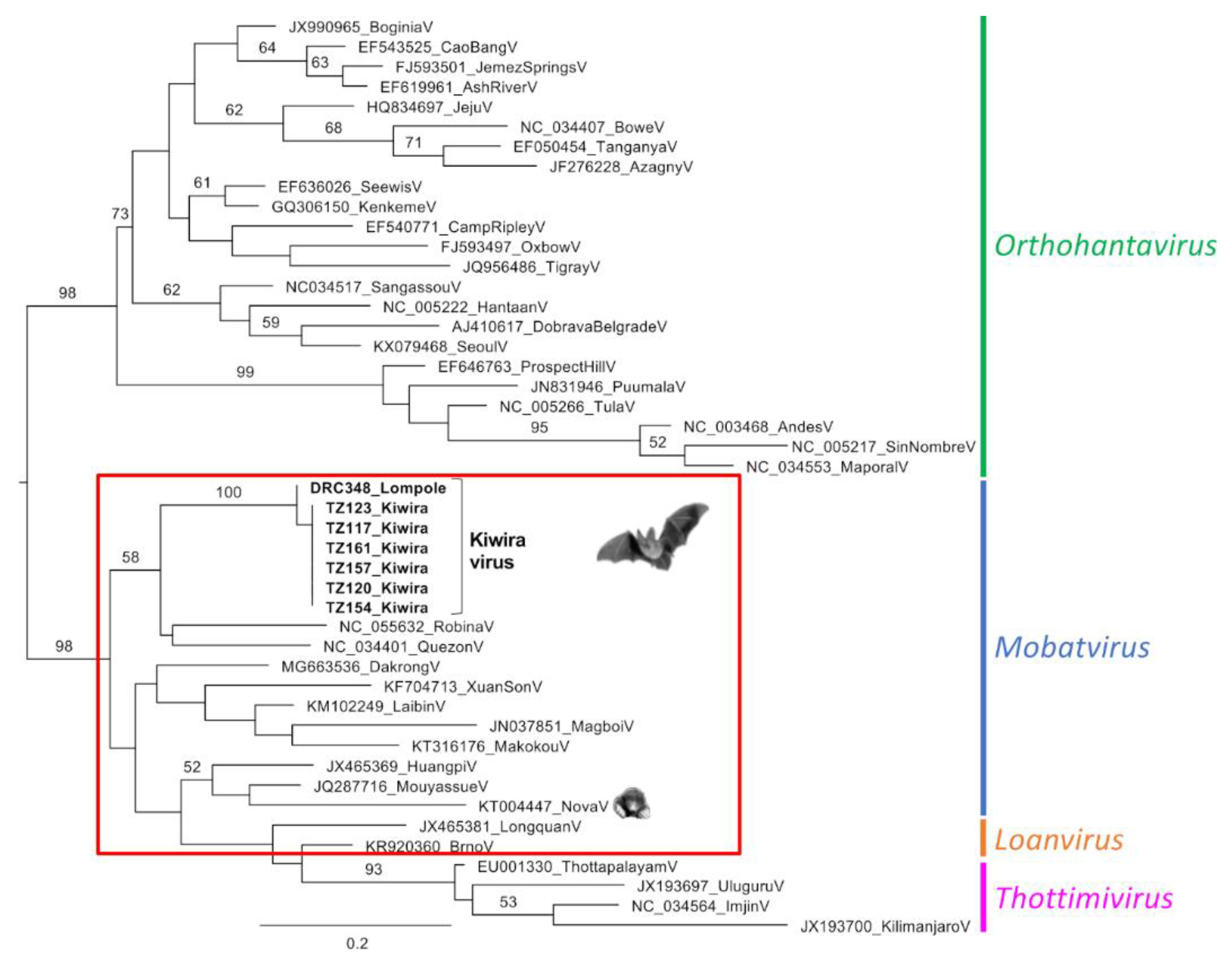
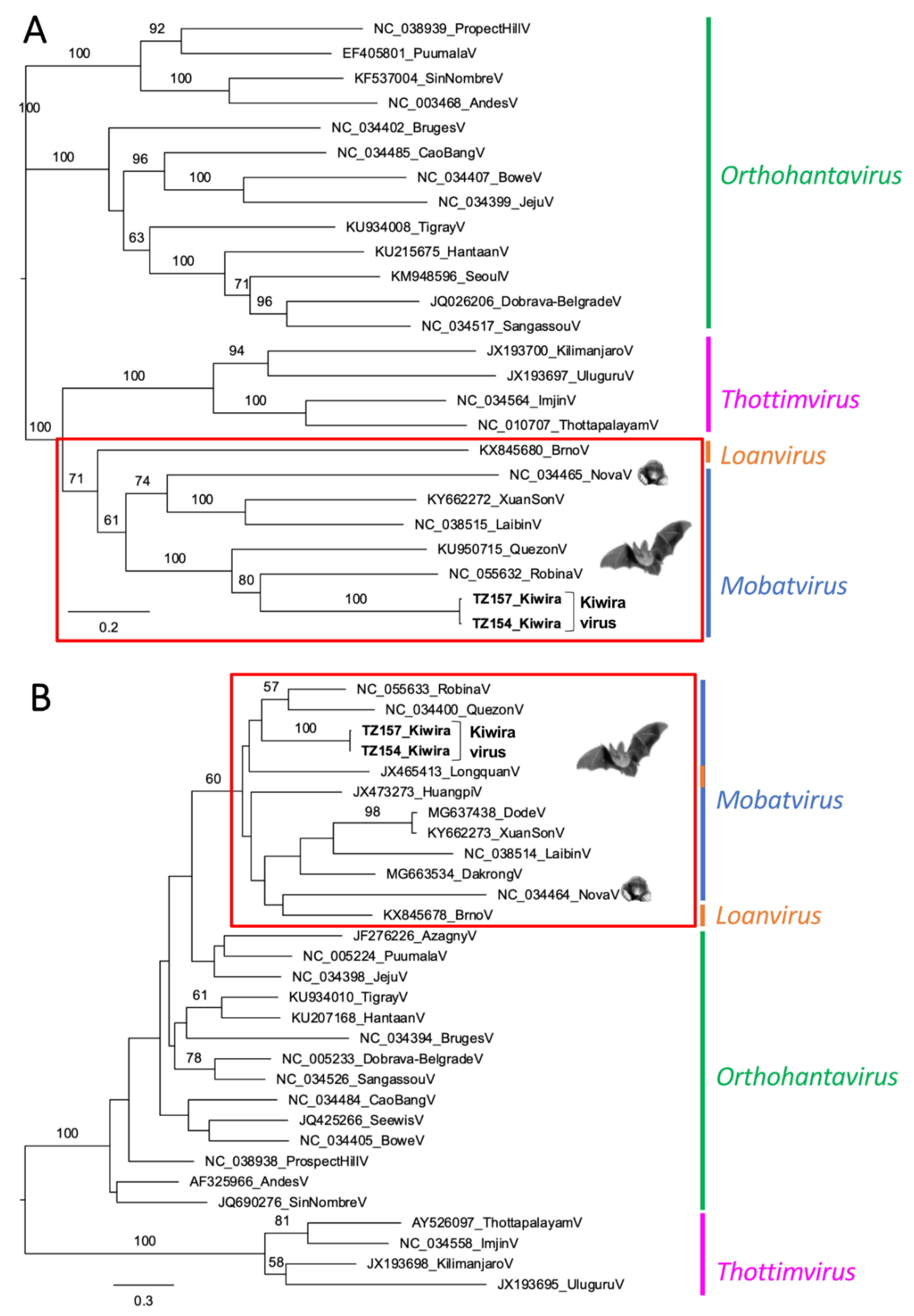
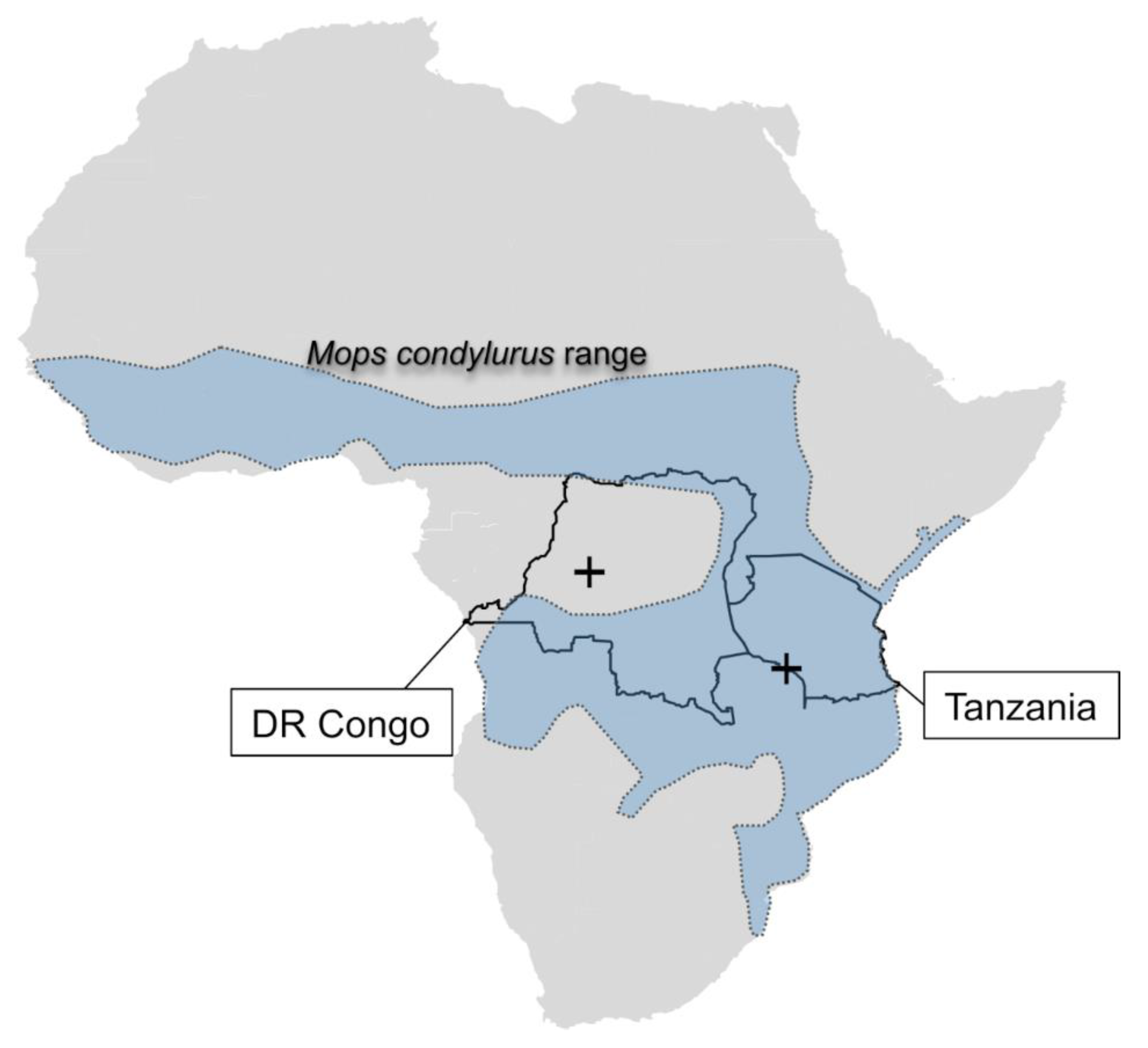
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vaheri, A.; Strandin, T.; Hepojoki, J.; Sironen, T.; Henttonen, H.; Mäkelä, S.; Mustonen, J. Uncovering the Mysteries of Hantavirus Infections. Nat. Rev. Microbiol. 2013, 11, 539–550. [Google Scholar] [CrossRef] [PubMed]
- Kruger, D.H.; Figueiredo, L.T.M.; Song, J.-W.; Klempa, B. Hantaviruses—Globally Emerging Pathogens. J. Clin. Virol. 2015, 64, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Denys, C.; Koivogui, L.; ter Meulen, J.; Krüger, D.H. Hantavirus in African Wood Mouse, Guinea. Emerg. Infect. Dis. 2006, 12, 838–840. [Google Scholar] [CrossRef] [PubMed]
- Klempa, B.; Fichet-Calvet, E.; Lecompte, E.; Auste, B.; Aniskin, V.; Meisel, H.; Barrière, P.; Koivogui, L.; ter Meulen, J.; Krüger, D.H. Novel Hantavirus Sequences in Shrew, Guinea. Emerg. Infect. Dis. 2007, 13, 520–522. [Google Scholar] [CrossRef]
- Weiss, S.; Witkowski, P.T.; Auste, B.; Nowak, K.; Weber, N.; Fahr, J.; Mombouli, J.-V.; Wolfe, N.D.; Drexler, J.F.; Drosten, C.; et al. Hantavirus in Bat, Sierra Leone. Emerg. Infect. Dis. 2012, 18, 159–161. [Google Scholar] [CrossRef]
- Sumibcay, L.; Kadjo, B.; Gu, S.H.; Kang, H.J.; Lim, B.K.; Cook, J.A.; Song, J.-W.; Yanagihara, R. Divergent Lineage of a Novel Hantavirus in the Banana Pipistrelle (Neoromicia nanus) in Côte d’Ivoire. Virol. J. 2012, 9, 34. [Google Scholar] [CrossRef]
- Klempa, B.; Koivogui, L.; Sylla, O.; Koulemou, K.; Auste, B.; Krüger, D.H.; ter Meulen, J. Serological Evidence of Human Hantavirus Infections in Guinea, West Africa. J. Infect. Dis. 2010, 201, 1031–1034. [Google Scholar] [CrossRef]
- Witkowski, P.T.; Leendertz, S.A.J.; Auste, B.; Akoua-Koffi, C.; Schubert, G.; Klempa, B.; Muyembe-Tamfum, J.-J.; Karhemere, S.; Leendertz, F.H.; Krüger, D.H. Human Seroprevalence Indicating Hantavirus Infections in Tropical Rainforests of Côte d’Ivoire and Democratic Republic of Congo. Front. Microbiol. 2015, 6, 518. [Google Scholar] [CrossRef]
- Heinemann, P.; Tia, M.; Alabi, A.; Anon, J.-C.; Auste, B.; Essbauer, S.; Gnionsahe, A.; Kigninlman, H.; Klempa, B.; Kraef, C.; et al. Human Infections by Non-Rodent-Associated Hantaviruses in Africa. J. Infect. Dis. 2016, 214, 1507–1511. [Google Scholar] [CrossRef]
- Arai, S.; Yanagihara, R. Genetic Diversity and Geographic Distribution of Bat-Borne Hantaviruses. Curr. Issues Mol. Biol. 2020, 39, 1–28. [Google Scholar] [CrossRef]
- Laenen, L.; Vergote, V.; Calisher, C.H.; Klempa, B.; Klingström, J.; Kuhn, J.H.; Maes, P. Hantaviridae: Current Classification and Future Perspectives. Viruses 2019, 11, 788. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning Sequence Reads, Clone Sequences and Assembly Contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar] [CrossRef]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Longueville, J.-E.; Gascuel, O. SMS: Smart Model Selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef]
- Whelan, S.; Goldman, N. A General Empirical Model of Protein Evolution Derived from Multiple Protein Families Using a Maximum-Likelihood Approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef]
- Schmidt, H.A.; Strimmer, K.; Vingron, M.; von Haeseler, A. TREE-PUZZLE: Maximum Likelihood Phylogenetic Analysis Using Quartets and Parallel Computing. Bioinformatics 2002, 18, 502–504. [Google Scholar] [CrossRef]
- Jones, K.E.; Purvis, A.; MacLarnon, A.; Bininda-Emonds, O.R.P.; Simmons, N.B. A Phylogenetic Supertree of the Bats (Mammalia: Chiroptera). Biol. Rev. Camb. Philos. Soc. 2002, 77, 223–259. [Google Scholar] [CrossRef]
- Arai, S.; Taniguchi, S.; Aoki, K.; Yoshikawa, Y.; Kyuwa, S.; Tanaka-Taya, K.; Masangkay, J.S.; Omatsu, T.; Puentespina, R.; Watanabe, S.; et al. Molecular Phylogeny of a Genetically Divergent Hantavirus Harbored by the Geoffroy’s Rousette (Rousettus amplexicaudatus), a Frugivorous Bat Species in the Philippines. Infect. Genet. Evol. 2016, 45, 26–32. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Adkins, S.; Agwanda, B.R.; Al Kubrusli, R.; Alkhovsky, S.V.; Amarasinghe, G.K.; Avšič-Županc, T.; Ayllón, M.A.; Bahl, J.; Balkema-Buschmann, A.; et al. 2021 Taxonomic Update of Phylum Negarnaviricota (Riboviria: Orthornavirae), Including the Large Orders Bunyavirales and Mononegavirales. Arch. Virol. 2021, 166, 3513–3566. [Google Scholar] [CrossRef] [PubMed]
- Witkowski, P.T.; Drexler, J.F.; Kallies, R.; Ličková, M.; Bokorová, S.; Maganga, G.D.; Szemes, T.; Leroy, E.M.; Krüger, D.H.; Drosten, C.; et al. Phylogenetic Analysis of a Newfound Bat-Borne Hantavirus Supports a Laurasiatherian Host Association for Ancestral Mammalian Hantaviruses. Infect. Genet. Evol. 2016, 41, 113–119. [Google Scholar] [CrossRef] [PubMed]
- Těšíková, J.; Bryjová, A.; Bryja, J.; Lavrenchenko, L.A.; Goüy de Bellocq, J. Hantavirus Strains in East Africa Related to Western African Hantaviruses. Vector Borne Zoonotic Dis. 2017, 17, 278–280. [Google Scholar] [CrossRef]
- Kariwa, H.; Fujiki, M.; Yoshimatsu, K.; Arikawa, J.; Takashima, I.; Hashimoto, N. Urine-Associated Horizontal Transmission of Seoul Virus among Rats. Arch. Virol. 1998, 143, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Safronetz, D.; Lindsay, R.; Dibernardo, A.; Hjelle, B.; Xiao, R.; Artsob, H.; Drebot, M.A. A Preliminary Study of the Patterns of Sin Nombre Viral Infection and Shedding in Naturally Infected Deer Mice (Peromyscus maniculatus). Vector Borne Zoonotic Dis. 2005, 5, 127–132. [Google Scholar] [CrossRef]
- Monadjem, A.; Cotterill, F.; Hutson, A.M.; Mickleburgh, S.; Bergmans, W. Mops condylurus. The IUCN Red List of Threatened Species 2017: E.T13838A22075340. Available online: https://doi.org/10.2305/IUCN.UK.2017-2.RLTS.T13838A22075340.en (accessed on 27 September 2022).
- Goldstein, T.; Anthony, S.J.; Gbakima, A.; Bird, B.H.; Bangura, J.; Tremeau-Bravard, A.; Belaganahalli, M.N.; Wells, H.L.; Dhanota, J.K.; Liang, E.; et al. The Discovery of Bombali Virus Adds Further Support for Bats as Hosts of Ebolaviruses. Nat. Microbiol. 2018, 3, 1084–1089. [Google Scholar] [CrossRef]
- Kareinen, L.; Ogola, J.; Kivistö, I.; Smura, T.; Aaltonen, K.; Jääskeläinen, A.J.; Kibiwot, S.; Masika, M.M.; Nyaga, P.; Mwaengo, D.; et al. Range Expansion of Bombali Virus in Mops condylurus Bats, Kenya, 2019. Emerg. Infect. Dis. 2020, 26, 3007–3010. [Google Scholar] [CrossRef]
- Kang, H.J.; Stanley, W.T.; Esselstyn, J.A.; Gu, S.H.; Yanagihara, R. Expanded Host Diversity and Geographic Distribution of Hantaviruses in Sub-Saharan Africa. J. Virol. 2014, 88, 7663–7667. [Google Scholar] [CrossRef]
- Guo, W.-P.; Lin, X.-D.; Wang, W.; Tian, J.-H.; Cong, M.-L.; Zhang, H.-L.; Wang, M.-R.; Zhou, R.-H.; Wang, J.-B.; Li, M.-H.; et al. Phylogeny and Origins of Hantaviruses Harbored by Bats, Insectivores, and Rodents. PLoS Pathog. 2013, 9, e1003159. [Google Scholar] [CrossRef]
- Arai, S.; Nguyen, S.T.; Boldgiv, B.; Fukui, D.; Araki, K.; Dang, C.N.; Ohdachi, S.D.; Nguyen, N.X.; Pham, T.D.; Boldbaatar, B.; et al. Novel Bat-Borne Hantavirus, Vietnam. Emerg. Infect. Dis. 2013, 19, 1159–1161. [Google Scholar] [CrossRef]
- Arai, S.; Kikuchi, F.; Bawm, S.; Sơn, N.T.; Lin, K.S.; Tú, V.T.; Aoki, K.; Tsuchiya, K.; Tanaka-Taya, K.; Morikawa, S.; et al. Molecular Phylogeny of Mobatviruses (Hantaviridae) in Myanmar and Vietnam. Viruses 2019, 11, 228. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sample ID | Sex | Location | GPS | Lung | Liver | Kidney | Spleen | Intestine |
|---|---|---|---|---|---|---|---|---|
| TZ117 | male | Kajunjumele | −9.609889, 33.913750 | + | - | - | - | NA |
| TZ120 | male | Kajunjumele | −9.609889, 33.913750 | + | + | NA | ||
| TZ123 | male | Kajunjumele | −9.609889, 33.913750 | NA | NA | |||
| TZ154 | male | Kyela | −9.602364, 33.925929 | + | + | + | ++ | + |
| TZ157 | female | Kyela | −9.602364, 33.925929 | ++ | + | + | +++ | + |
| TZ161 | male | Kyela | −9.602364, 33.925929 | |||||
| DRC348 | NA | Lompole | −2.5881744, 20.3696069 | NA | NA | NA | NA |
| NC_034401 Quezon Virus | NC_005632 Robina Virus | DRC348 Lompole | |
|---|---|---|---|
| TZ117 | 82.1 | 80.0 | 98.6 |
| TZ120 | 81.4 | 80.0 | 98.6 |
| TZ123 | 82.1 | 80.7 | 98.6 |
| TZ154 | 82.1 | 80.0 | 98.6 |
| TZ157 | 82.1 | 80.7 | 98.6 |
| TZ161 | 82.1 | 80.7 | 98.6 |
| DRC348 | 82.9 | 81.4 |
| Kiwira Virus Samples | Pairwise Identity [%] to Quezon Virus 2 | Pairwise Identity [%] to Robina Virus 3 | Number of Mapped Reads |
|---|---|---|---|
| TZ154S | 87.4 | 88.3 | 39,234 |
| TZ157S | 85.4 | 86.4 | 2480 |
| TZ154L | 82.9 | 83.7 | 1,997,514 |
| TZ157L | 80.4 | 81.6 | 1,478,369 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weiss, S.; Sudi, L.E.; Düx, A.; Mangu, C.D.; Ntinginya, N.E.; Shirima, G.M.; Köndgen, S.; Schubert, G.; Witkowski, P.T.; Muyembe, J.-J.; et al. Kiwira Virus, a Newfound Hantavirus Discovered in Free-tailed Bats (Molossidae) in East and Central Africa. Viruses 2022, 14, 2368. https://doi.org/10.3390/v14112368
Weiss S, Sudi LE, Düx A, Mangu CD, Ntinginya NE, Shirima GM, Köndgen S, Schubert G, Witkowski PT, Muyembe J-J, et al. Kiwira Virus, a Newfound Hantavirus Discovered in Free-tailed Bats (Molossidae) in East and Central Africa. Viruses. 2022; 14(11):2368. https://doi.org/10.3390/v14112368
Chicago/Turabian StyleWeiss, Sabrina, Lwitiho E. Sudi, Ariane Düx, Chacha D. Mangu, Nyanda Elias Ntinginya, Gabriel M. Shirima, Sophie Köndgen, Grit Schubert, Peter T. Witkowski, Jean-Jacques Muyembe, and et al. 2022. "Kiwira Virus, a Newfound Hantavirus Discovered in Free-tailed Bats (Molossidae) in East and Central Africa" Viruses 14, no. 11: 2368. https://doi.org/10.3390/v14112368
APA StyleWeiss, S., Sudi, L. E., Düx, A., Mangu, C. D., Ntinginya, N. E., Shirima, G. M., Köndgen, S., Schubert, G., Witkowski, P. T., Muyembe, J.-J., Ahuka, S., Klempa, B., Leendertz, F. H., & Krüger, D. H. (2022). Kiwira Virus, a Newfound Hantavirus Discovered in Free-tailed Bats (Molossidae) in East and Central Africa. Viruses, 14(11), 2368. https://doi.org/10.3390/v14112368