
Structural Insights into Common and Host-Specific Receptor-Binding Mechanisms in Algal Picorna-like Viruses


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Propagation and Sample Purification
2.2. Cryo-EM Data Collection and Single Particle Analysis
2.3. Atomic Modeling and Refinement
2.4. Structural Analysis
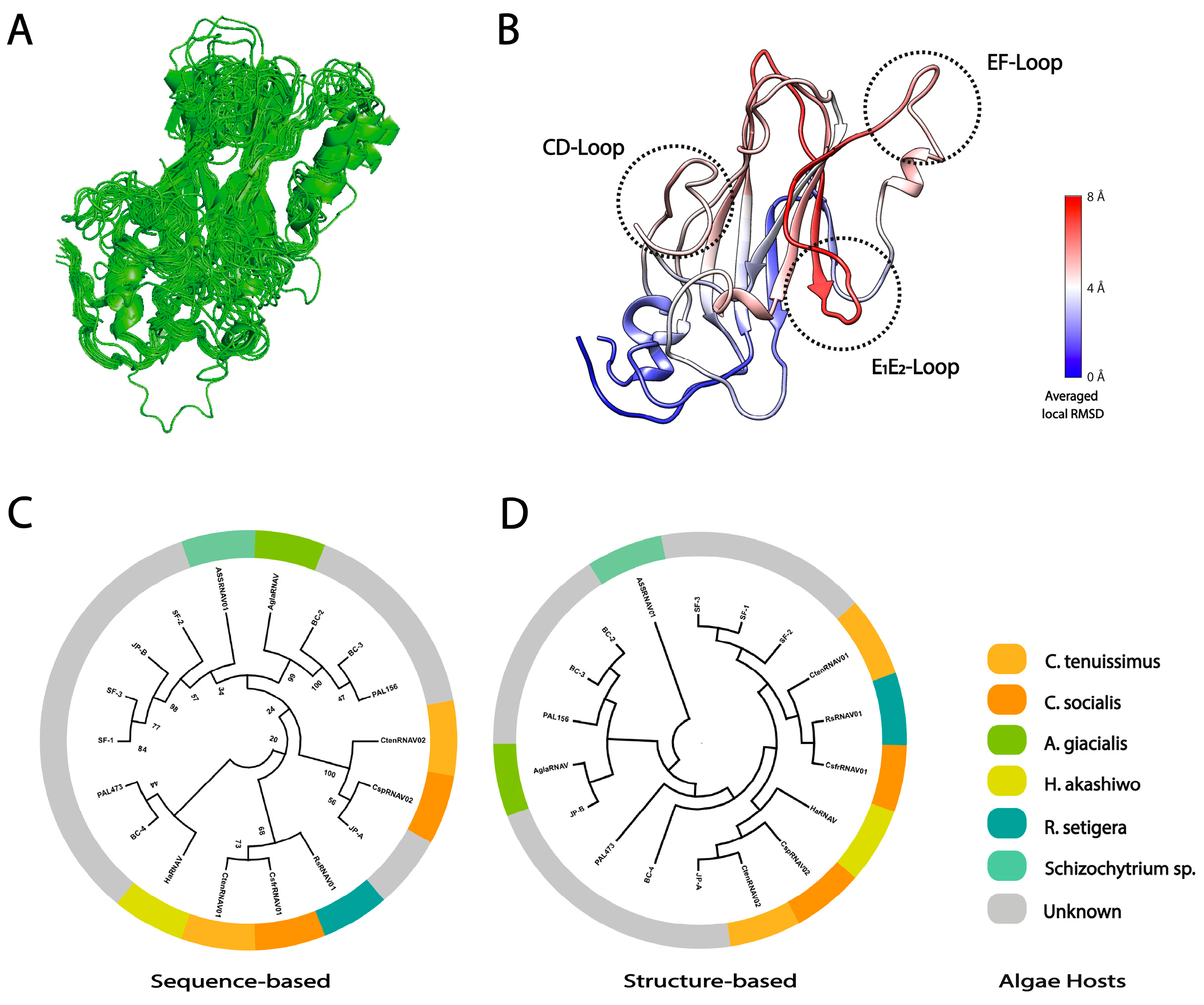
2.5. Sequence- and Structure-Based Phylogeny
3. Results and Discussion
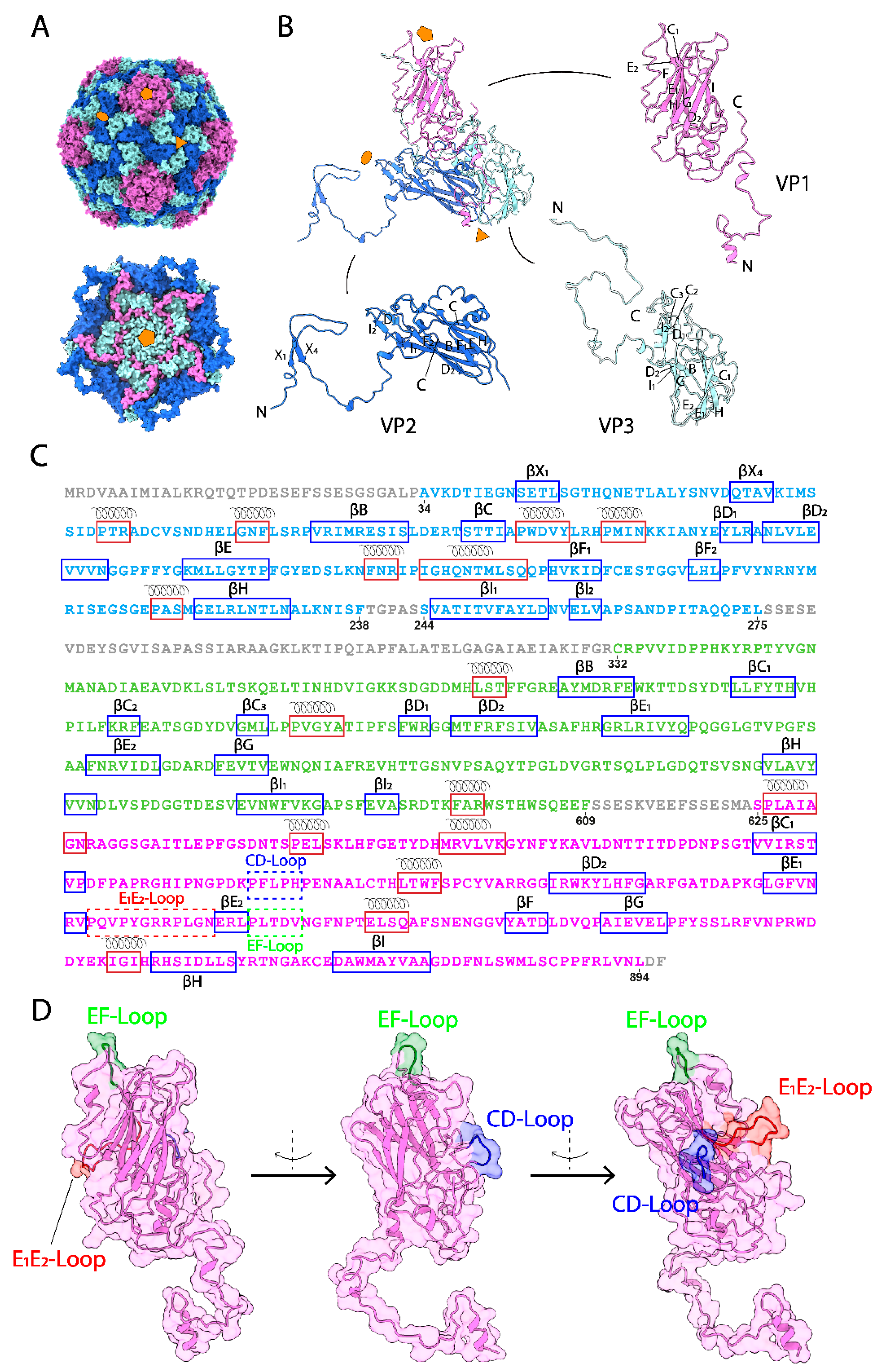
3.1. The Overall Capsid Structure of CsfrRNAV and Its Capsid Proteins
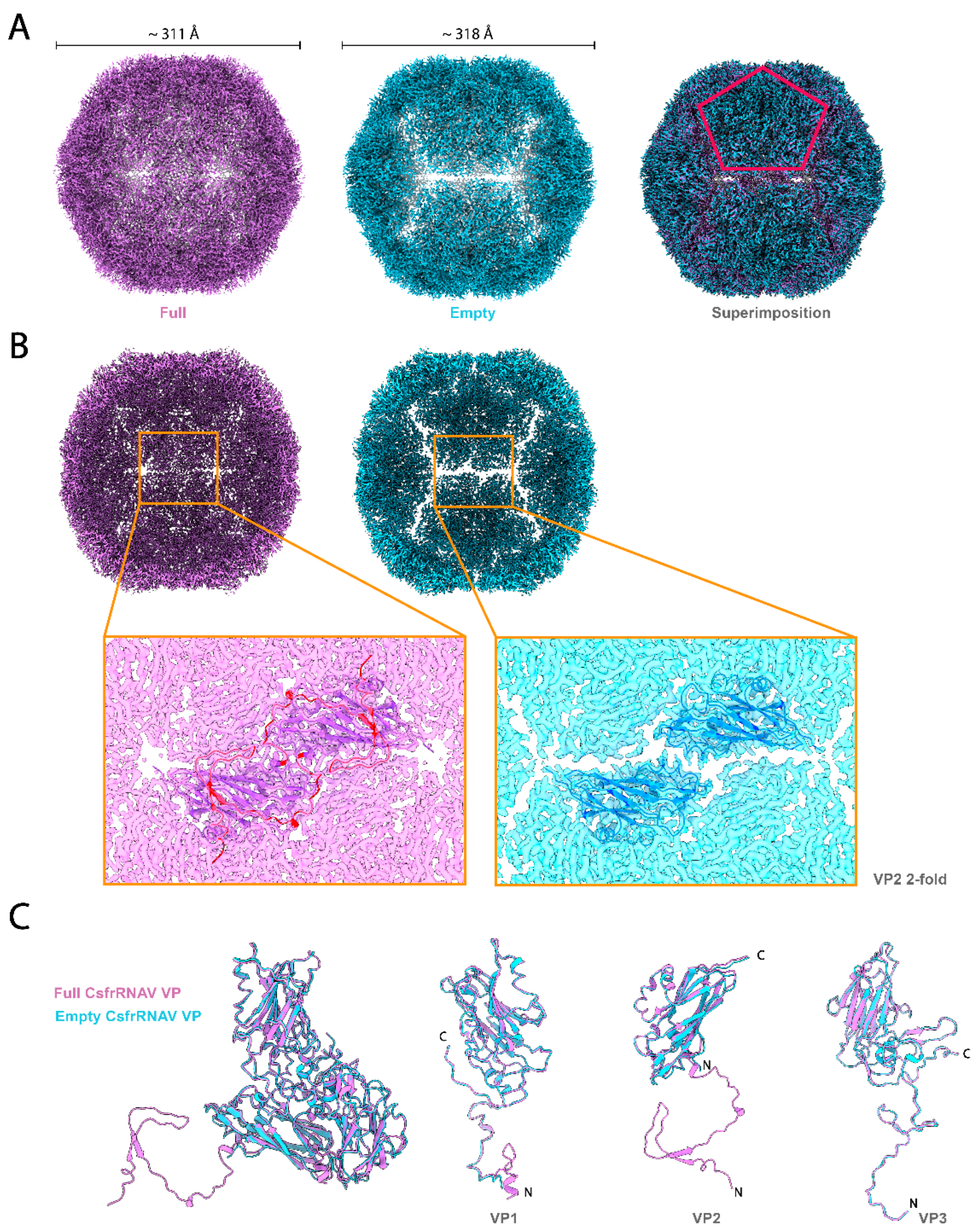
3.2. Empty Particle Structure and Variability
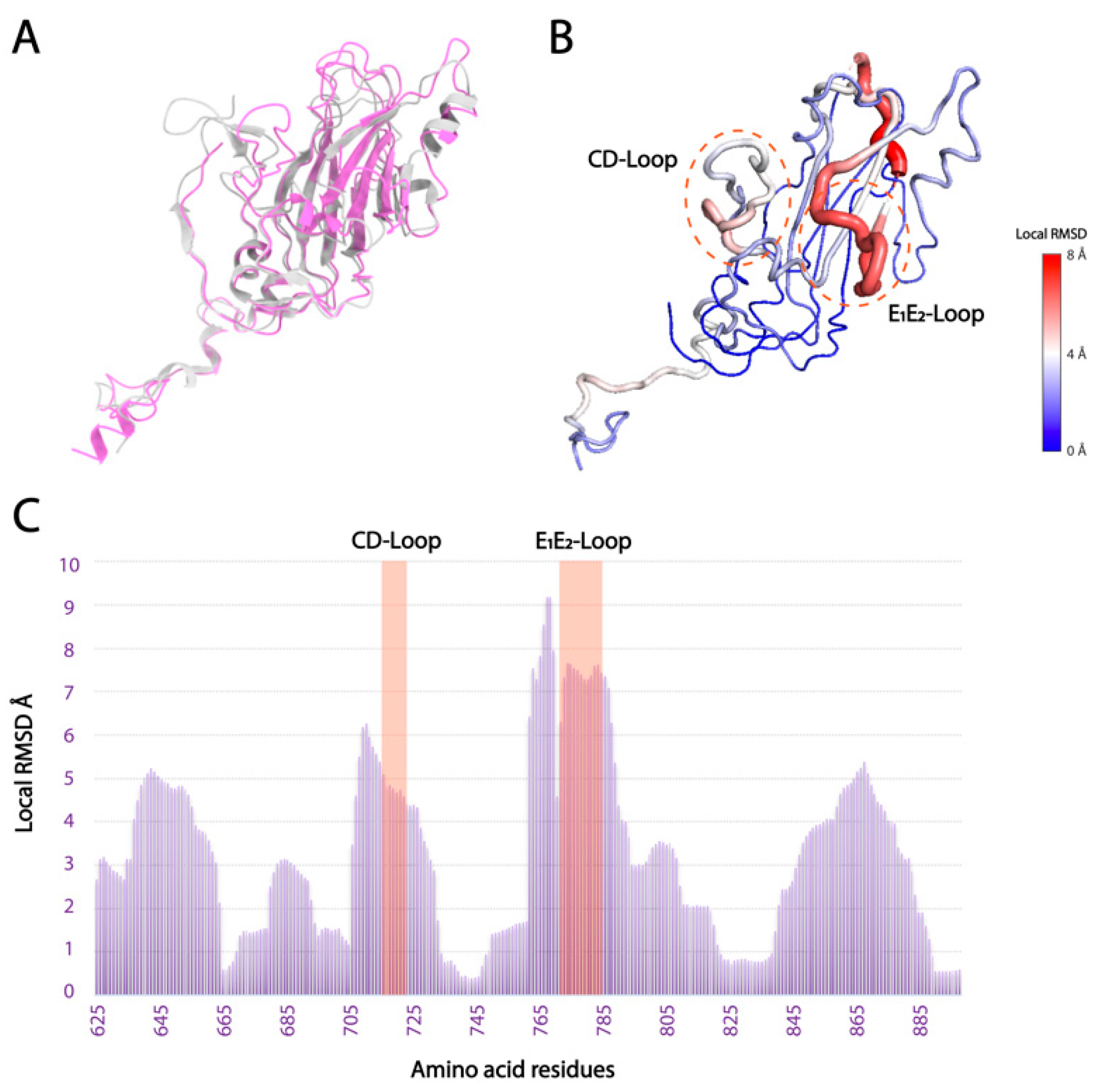
3.3. Local Structural Differences of VP1 in Two Marnaviridae Viruses
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Suttle, C.A. Marine viruses—Major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Brussaard, C.P.D. Viral Control of Phytoplankton Populations—A Review. J. Eukaryot. Microbiol. 2004, 51, 125–138. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, T.V.; Larsen, A.; Bratbak, G.; Pagarete, A.; Edvardsen, B.; Egge, E.D.; Sandaa, R.A. Seasonal dynamics of haptophytes and dsDNA algal viruses suggest complex virus-host relationship. Viruses 2017, 9, 84. [Google Scholar] [CrossRef]
- Sadeghi, M.; Tomaru, Y.; Ahola, T. RNA viruses in aquatic unicellular eukaryotes. Viruses 2021, 13, 362. [Google Scholar] [CrossRef]
- Coy, S.R.; Gann, E.R.; Pound, H.L.; Short, S.M.; Wilhelm, S.W. Viruses of eukaryotic algae: Diversity, methods for detection, and future directions. Viruses 2018, 10, 487. [Google Scholar] [CrossRef] [PubMed]
- Vlok, M.; Lang, A.S.; Suttle, C.A. Marine RNA Virus Quasispecies Are Distributed throughout the Oceans. mSphere 2019, 4, 1–18. [Google Scholar] [CrossRef]
- Zell, R.; Groth, M.; Selinka, L.; Selinka, H.C. Picorna-Like Viruses of the Havel River, Germany. Front. Microbiol. 2022, 13, 865287. [Google Scholar] [CrossRef]
- Tai, V.; Lawrence, J.E.; Lang, A.S.; Chan, A.M.; Culley, A.I.; Suttle, C.A. Characterization of HaRNAV, a single-stranded RNA virus causing lysis of Heterosigma Akashiwo. J. Phycol. 2003, 39, 343–352. [Google Scholar] [CrossRef]
- Nagasaki, K.; Tomaru, Y.; Katanozaka, N.; Shirai, Y.; Nishida, K.; Itakura, S.; Yamaguchi, M. Isolation and characterization of a novel single-stranded RNA virus infecting the bloom-forming diatom Rhizosolenia setigera. Appl. Environ. Microbiol. 2004, 70, 704–711. [Google Scholar] [CrossRef]
- Shirai, Y.; Tomaru, Y.; Takao, Y.; Suzuki, H.; Nagumo, T.; Nagasaki, K. Isolation and characterization of a single-stranded RNA virus infecting the marine planktonic diatom Chaetoceros tenuissimus meunier. Appl. Environ. Microbiol. 2008, 74, 4022–4027. [Google Scholar] [CrossRef]
- Tomaru, Y.; Takao, Y.; Suzuki, H.; Nagumo, T.; Nagasaki, K. Isolation and characterization of a single-stranded RNA virus infecting the bloom-forming diatom Chaetoceros socialis. Appl. Environ. Microbiol. 2009, 75, 2375–2381. [Google Scholar] [CrossRef]
- Tomaru, Y.; Toyoda, K.; Kimura, K.; Takao, Y.; Sakurada, K.; Nakayama, N.; Nagasaki, K. Isolation and characterization of a single-stranded RNA virus that infects the marine planktonic diatom Chaetoceros sp. (SS08-C03). Phycol. Res. 2013, 61, 27–36. [Google Scholar] [CrossRef]
- Kimura, K.; Tomaru, Y. Discovery of two novel viruses expands the diversity of single-stranded DNA and single-stranded RNA viruses infecting a cosmopolitan marine diatom. Appl. Environ. Microbiol. 2015, 81, 1120–1131. [Google Scholar] [CrossRef] [PubMed]
- Lang, A.S.; Vlok, M.; Culley, A.; Suttle, C.; Takao, Y.; Tomaru, Y.; Siddell, S.; Lefkowitz, E.; Simmonds, P.; Zerbini, F.; et al. ICTV virus taxonomy profile: Marnaviridae 2021. J. Gen. Virol. 2021, 102, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Munke, A.; Kimura, K.; Tomaru, Y.; Okamoto, K. Capsid Structure of a Marine Algal Virus of the Order Picornavirales. J. Virol. 2020, 94, e01855-19. [Google Scholar] [CrossRef]
- Rossmann, M.G.; Arnold, E.; Erickson, J.W.; Frankenberger, E.A.; Griffith, J.P.; Hecht, H.J.; Johnson, J.E.; Kamer, G.; Luo, M.; Mosser, A.G.; et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 1985, 317, 145–153. [Google Scholar] [CrossRef]
- Rossmann, M.G. The Canyon Hypothesis. J. Biol. Chem. 1989, 264, 14587–14590. [Google Scholar] [CrossRef]
- Rossmann, M.G.; He, Y.; Kuhn, R.J. Picornavirus-receptor interactions. Trends Microbiol. 2002, 10, 324–331. [Google Scholar] [CrossRef]
- Zocher, G.; Mistry, N.; Frank, M.; Schick, I.H.; Ekström, J.O.; Arnberg, N.; Stehle, T. A Sialic Acid Binding Site in a Human Picornavirus. PLoS Pathog. 2014, 10, e1004401. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, X.; Ren, J.; Kotecha, A.; Walter, T.S.; Yuan, S.; Yamashita, T.; Tuthill, T.J.; Fry, E.E.; Rao, Z.; et al. Structure of human Aichi virus and implications for receptor binding. Nat. Microbiol. 2016, 1, 1–6. [Google Scholar] [CrossRef]
- Zhu, L.; Wang, X.; Ren, J.; Porta, C.; Wenham, H.; Ekström, J.O.; Panjwani, A.; Knowles, N.J.; Kotecha, A.; Siebert, C.A.; et al. Structure of Ljungan virus provides insight into genome packaging of this picornavirus. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Levy, H.C.; Bostina, M.; Filman, D.J.; Hogle, J.M. Catching a Virus in the Act of RNA Release: A Novel Poliovirus Uncoating Intermediate Characterized by Cryo-Electron Microscopy. J. Virol. 2010, 84, 4426–4441. [Google Scholar] [CrossRef] [PubMed]
- Bostina, M.; Levy, H.; Filman, D.J.; Hogle, J.M. Poliovirus RNA Is Released from the Capsid near a Twofold Symmetry Axis. J. Virol. 2011, 85, 776–783. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Peng, W.; Ren, J.; Hu, Z.; Xu, J.; Lou, Z.; Li, X.; Yin, W.; Shen, X.; Porta, C.; et al. A sensor-adaptor mechanism for enterovirus uncoating from structures of EV71. Nat. Struct Mol. Biol. 2012, 19, 424–429. [Google Scholar] [CrossRef] [PubMed]
- Seitsonen, J.J.T.; Shakeel, S.; Susi, P.; Pandurangan, A.P.; Sinkovits, R.S.; Hyvönen, H.; Laurinmäki, P.; Pelto, J.Y.; Topf, M.; Hyypiä, T.; et al. Structural Analysis of Coxsackievirus A7 Reveals Conformational Changes Associated with Uncoating. J. Virol. 2012, 86, 7207–7215. [Google Scholar] [CrossRef]
- Garriga, D.; Herk, A.P.; Luque, D.; Wruss, J.; Castón, J.R.; Blaas, D.; Verdaguer, N. Insights into minor group rhinovirus uncoating: The X-ray structure of the HRV2 empty capsid. PLoS Pathog. 2012, 8, e1002473. [Google Scholar] [CrossRef]
- Ren, J.; Wang, X.; Hu, Z.; Gao, Q.; Yao, S.; Li, X.; Porta, C.; Walter, T.S.; Gilber, R.J.; Zhao, Y.; et al. Picornavirus uncoating intermediate captured in atomic detail. Nat. Commun. 2013, 4, 1–7. [Google Scholar] [CrossRef]
- Shingler, K.L.; Yoder, J.L.; Carnegie, M.S.; Ashley, R.E.; Makhov, A.M.; Conway, J.F.; Hafenstein, S. The Enterovirus 71 A-particle Forms a Gateway to Allow Genome Release: A CryoEM Study of Picornavirus Uncoating. PLoS Pathog. 2013, 9, e1003240. [Google Scholar] [CrossRef]
- Lyu, K.; Ding, J.; Han, J.; Zhang, Y.; Wu, X.; He, Y.; Qin, C.; Chen, R. Human Enterovirus 71 Uncoating Captured at Atomic Resolution. J. Virol. 2014, 88, 3114–3126. [Google Scholar] [CrossRef]
- Lee, H.; Shingler, K.L.; Organtini, L.J.; Ashley, R.E.; Makhov, A.M.; Conway, J.F.; Hafenstein, S. The novel asymmetric entry intermediate of a picornavirus captured with nanodiscs. Sci. Adv. 2016, 2, e1501929. [Google Scholar] [CrossRef]
- Sabin, C.; Füzik, T.; Škubník, K.; Pálková, L.; Lindberg, A.M.; Plevka, P. Structure of Aichi Virus 1 and Its Empty Particle: Clues to Kobuvirus Genome Release Mechanism. J. Virol. 2016, 90, 10800–10810. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Sun, Y.; Fan, J.; Zhu, B.; Cao, L.; Gao, Q.; Zhang, Y.; Liu, H.; Rao, Z.; Wang, X. Structures of Coxsackievirus A10 unveil the molecular mechanisms of receptor binding and viral uncoating. Nat. Commun. 2018, 9, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Yafal, A.G.; Lee, Y.M.; Hogle, J.; Chow, M. Poliovirus neutralization by antibodies to internal epitopes of VP4 and VP1 results from reversible exposure of these sequences at physiological temperature. J. Virol. 1994, 68, 3965–3970. [Google Scholar] [CrossRef] [PubMed]
- Katpally, U.; Fu, T.-M.; Freed, D.C.; Casimiro, D.R.; Smith, T.J. Antibodies to the Buried N Terminus of Rhinovirus VP4 Exhibit Cross-Serotypic Neutralization. J. Virol. 2009, 83, 7040–7048. [Google Scholar] [CrossRef]
- Panjwani, A.; Asfor, A.S.; Tuthill, T.J. The conserved N-terminus of human rhinovirus capsid protein VP4 contains membrane pore-forming activity and is a target for neutralizing antibodies. J. Gen. Virol. 2016, 97, 3238–3242. [Google Scholar] [CrossRef]
- Kelly, J.T.; Swanson, J.; Newman, J.; Groppelli, E.; Stonehouse, N.J.; Tuthill, T.J. Membrane Interactions and Uncoating of Aichi Virus, a Picornavirus That Lacks a VP4. J. Virol. 2022, 96, e00082-22. [Google Scholar] [CrossRef]
- Bakker, S.E.; Groppelli, E.; Pearson, A.R.; Stockley, P.G.; Rowlands, D.J.; Ranson, N.A. Limits of Structural Plasticity in a Picornavirus Capsid Revealed by a Massively Expanded Equine Rhinitis A Virus Particle. J. Virol. 2014, 88, 6093–6099. [Google Scholar] [CrossRef]
- Punjani, A.; Rubinstein, J.L.; Fleet, D.J.; Brubaker, M.A. CryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 2017, 14, 290–296. [Google Scholar] [CrossRef] [PubMed]
- Punjani, A.; Fleet, D.J. 3D variability analysis: Resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM. J. Struct. Biol. 2021, 213, 107702. [Google Scholar] [CrossRef]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef]
- Liebschner, D.; Afonine, P.V.; Baker, M.L.; Bunkóczi, G.; Chen, V.B.; Croll, T.I.; Hintze, B.; Hung, L.; Jain, S.; McCoy, A.J.; et al. Macromolecular structure determination using X-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. Sect. D Biol. Crystallogr. 2019, 75, 861–877. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure visualization for researchers, educators, and developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar] [CrossRef]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Varadi, M.; Anyango, S.; Deshpande, M.; Nair, S.; Natassia, C.; Yordanova, G.; Yuan, D.; Stroe, O.; Wood, G.; Laydon, A.; et al. AlphaFold Protein Structure Database: Massively expanding the structural coverage of protein-sequence space with high-accuracy models. Nucleic Acids Res. 2022, 50, D439–D444. [Google Scholar] [CrossRef]
- Konagurthu, A.S.; Whisstock, J.C.; Stuckey, P.J.; Lesk, A.M. MUSTANG: A multiple structural alignment algorithm. Proteins 2006, 64, 559–574. [Google Scholar] [CrossRef]
- Munke, A.; Kimura, K.; Tomaru, Y.; Wang, H.; Yoshida, K.; Mito, S.; Hongo, Y.; Okamoto, K. Primordial Capsid and Spooled ssDNA Genome Structures Unravel Ancestral Events of Eukaryotic Viruses. mBio 2022, 13, e00156-22. [Google Scholar] [CrossRef]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis Version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef]
- Wang, X.; Ren, J.; Gao, Q.; Hu, Z.; Sun, Y.; Li, X.; Rowlands, D.J.; Yin, W.; Wang, J.; Stuart, D.I.; et al. Hepatitis A virus and the origins of picornaviruses. Nature 2015, 517, 85–88. [Google Scholar] [CrossRef]
- Li, C.; Wang, J.C.-Y.; Taylor, M.W.; Zlotnick, A. In Vitro Assembly of an Empty Picornavirus Capsid follows a Dodecahedral Path. J. Virol. 2012, 86, 13062–13069. [Google Scholar] [CrossRef]
- Chandler-Bostock, R.; Mata, C.P.; Bingham, R.J.; Dykeman, E.C.; Meng, B.; Tuthill, T.J.; Rowlands, D.J.; Ranson, N.A.; Twarock, R.; Stockley, P.G. Assembly of infectious enteroviruses depends on multiple, conserved genomic RNA-coat protein contacts. PLoS Pathog. 2020, 16, e1009146. [Google Scholar] [CrossRef]
- Buchta, D.; Füzik, T.; Hrebík, D.; Levdansky, Y.; Sukeník, L.; Mukhamedova, L.; Moravcová, J.; Vácha, R.; Plevka, P. Enterovirus particles expel capsid pentamers to enable genome release. Nat. Commun. 2019, 10, 1–9. [Google Scholar] [CrossRef]
- Škubník, K.; Sukeník, L.; Buchta, D.; Füzik, T.; Procházková, M.; Moravcová, J.; Šmerdová, L.; Přidal, A.; Vácha, R.; Plevka, P. Capsid opening enables genome release of iflaviruses. Sci. Adv. 2021, 7, eabd7130. [Google Scholar] [CrossRef]
- Hrebík, D.; Füzik, T.; Gondová, M.; Šmerdová, L.; Adamopoulos, A.; Šedo, O.; Zdráhal, Z.; Plevka, P. ICAM-1 induced rearrangements of capsid and genome prime rhinovirus 14 for activation and uncoating. Proc. Natl. Acad. Sci. USA 2021, 118, e2024251118. [Google Scholar] [CrossRef]
- Cifuente, J.O.; Lee, H.; Yoder, J.D.; Shingler, K.L.; Carnegie, M.S.; Yoder, J.L.; Ashley, R.E.; Makhov, A.M.; Conway, J.F.; Hafenstein, S. Structures of the Procapsid and Mature Virion of Enterovirus 71 Strain 1095. J. Virol. 2013, 87, 7637–7645. [Google Scholar] [CrossRef]
- Wang, H.; Salaipeth, L.; Miyazaki, N.; Suzuki, N.; Okamoto, K. Capsid structure of a metazoan fungal dsRNA megabirnavirus reveals its uniquely acquired structures. bioRxiv 2022. [Google Scholar] [CrossRef]
- Tommaso, P.D.; Moretti, S.; Xenarios, I.; Orobitg, M.; Montanyola, A.; Chang, J.; Taly, J.; Notredame, C. T-Coffee: A web server for the multiple sequence alignment of protein and RNA sequences using structural information and homology extension. Nucleic Acids Res. 2011, 39, 13–17. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Munke, A.; Li, S.; Tomaru, Y.; Okamoto, K. Structural Insights into Common and Host-Specific Receptor-Binding Mechanisms in Algal Picorna-like Viruses. Viruses 2022, 14, 2369. https://doi.org/10.3390/v14112369
Wang H, Munke A, Li S, Tomaru Y, Okamoto K. Structural Insights into Common and Host-Specific Receptor-Binding Mechanisms in Algal Picorna-like Viruses. Viruses. 2022; 14(11):2369. https://doi.org/10.3390/v14112369
Chicago/Turabian StyleWang, Han, Anna Munke, Siqi Li, Yuji Tomaru, and Kenta Okamoto. 2022. "Structural Insights into Common and Host-Specific Receptor-Binding Mechanisms in Algal Picorna-like Viruses" Viruses 14, no. 11: 2369. https://doi.org/10.3390/v14112369
APA StyleWang, H., Munke, A., Li, S., Tomaru, Y., & Okamoto, K. (2022). Structural Insights into Common and Host-Specific Receptor-Binding Mechanisms in Algal Picorna-like Viruses. Viruses, 14(11), 2369. https://doi.org/10.3390/v14112369