
Evolutionary Signatures Governing the Codon Usage Bias in Coronaviruses and Their Implications for Viruses Infecting Various Bat Species

 ,
,  ,
,  , and
, and 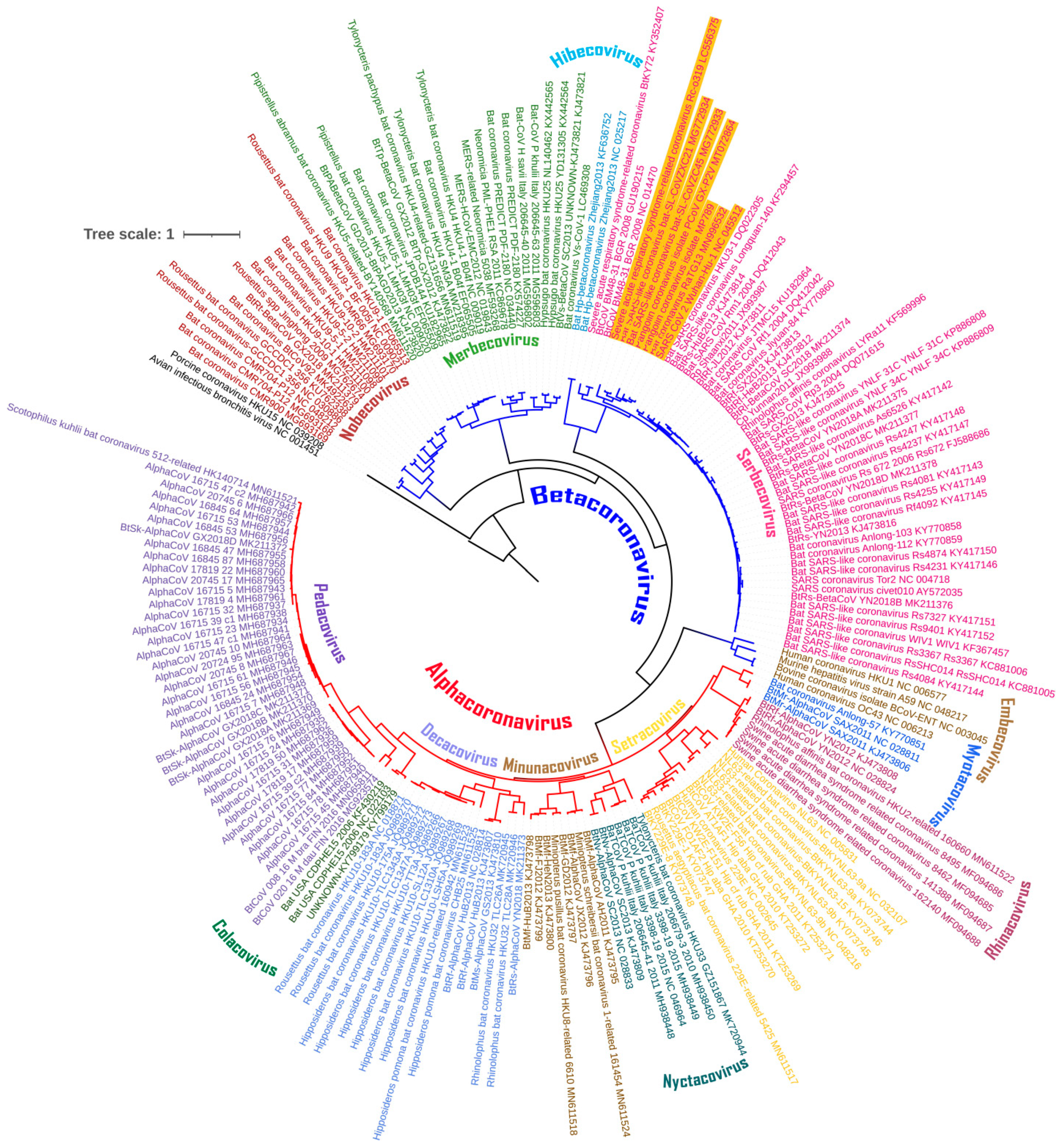{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Coronavirus Genome Data Set
2.2. Maximum-Likelihood Analysis
2.3. Dinucleotide Composition Analysis
2.4. Relative Synonymous Codon Usage Analysis
2.5. Statistical Analysis
3. Results
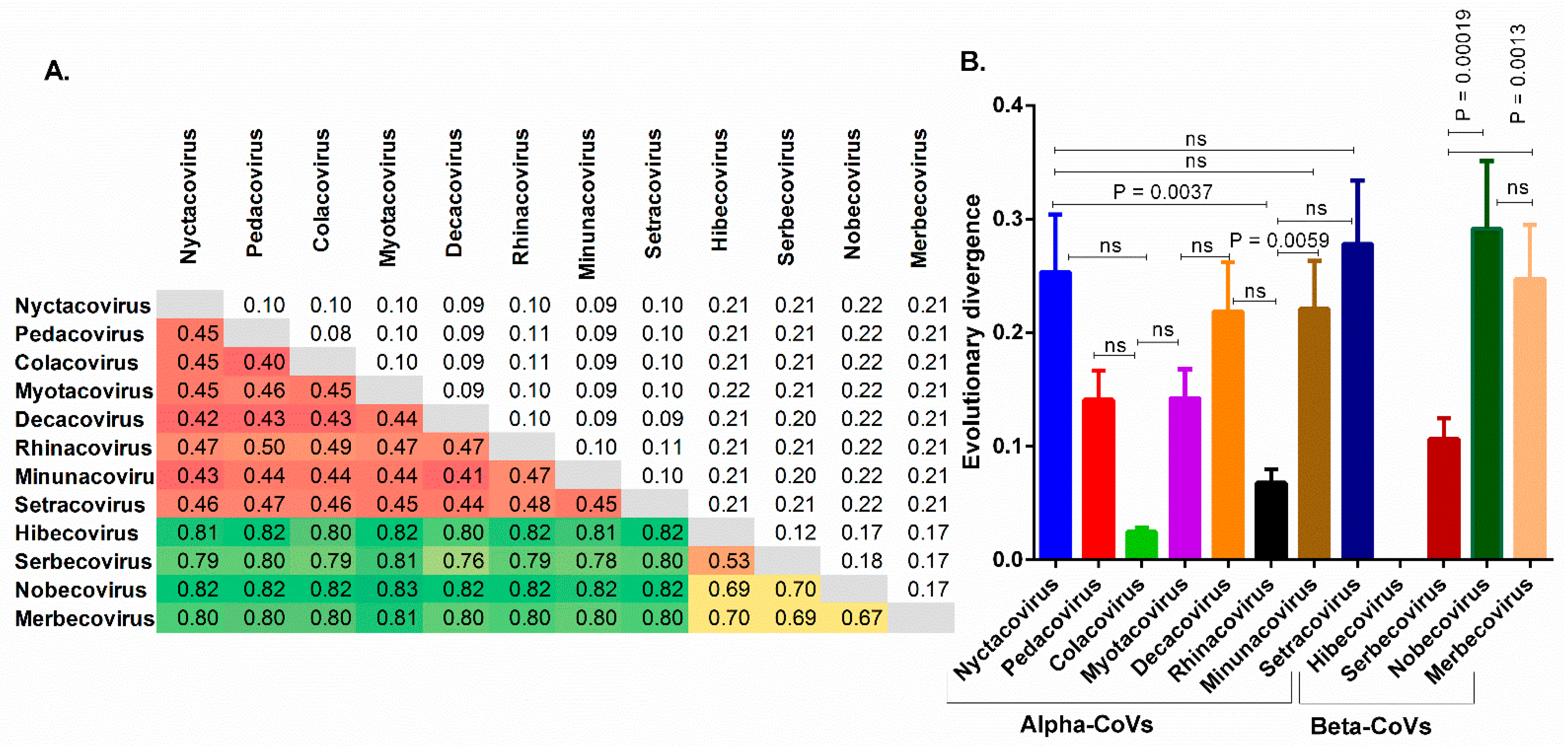
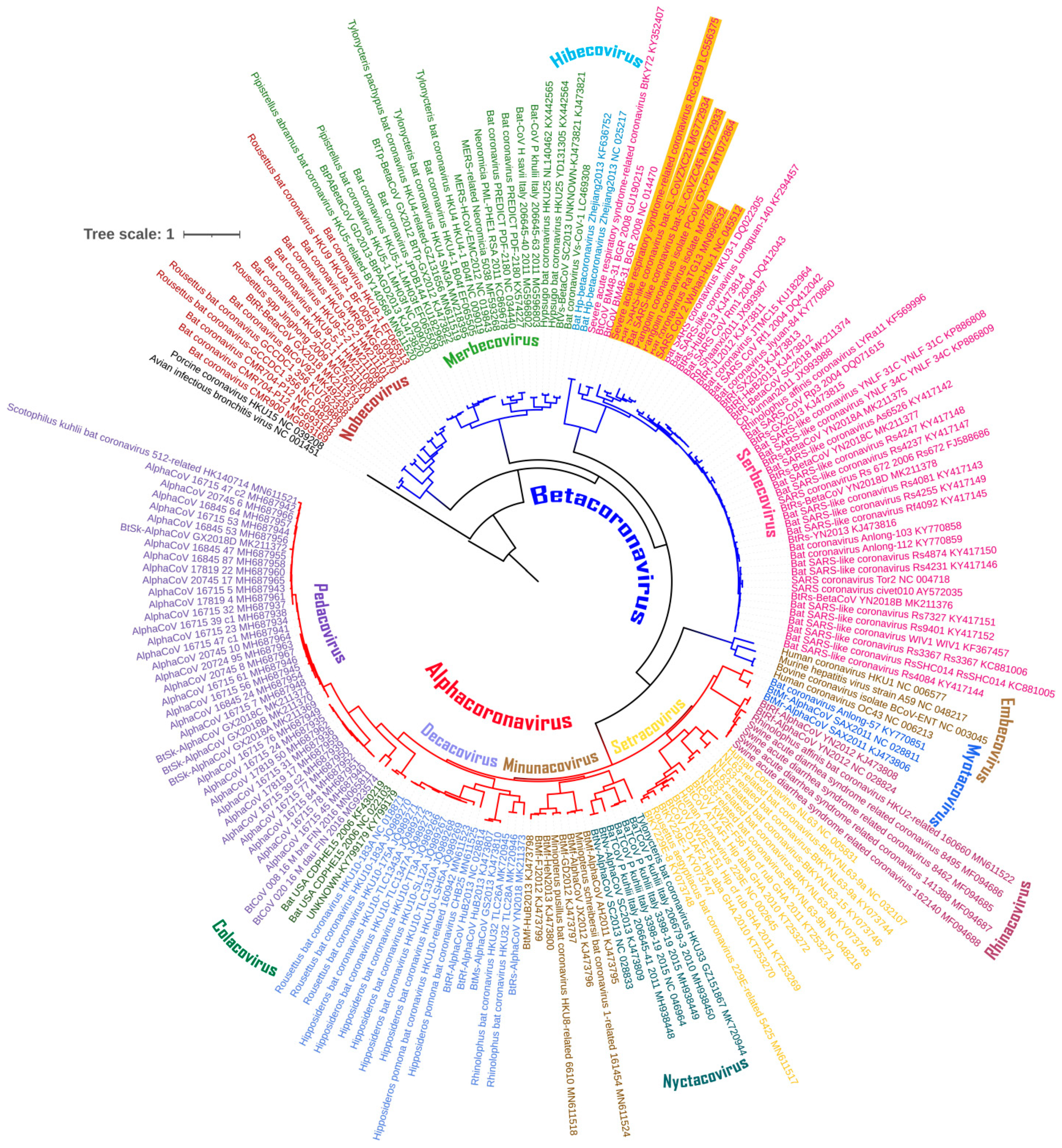
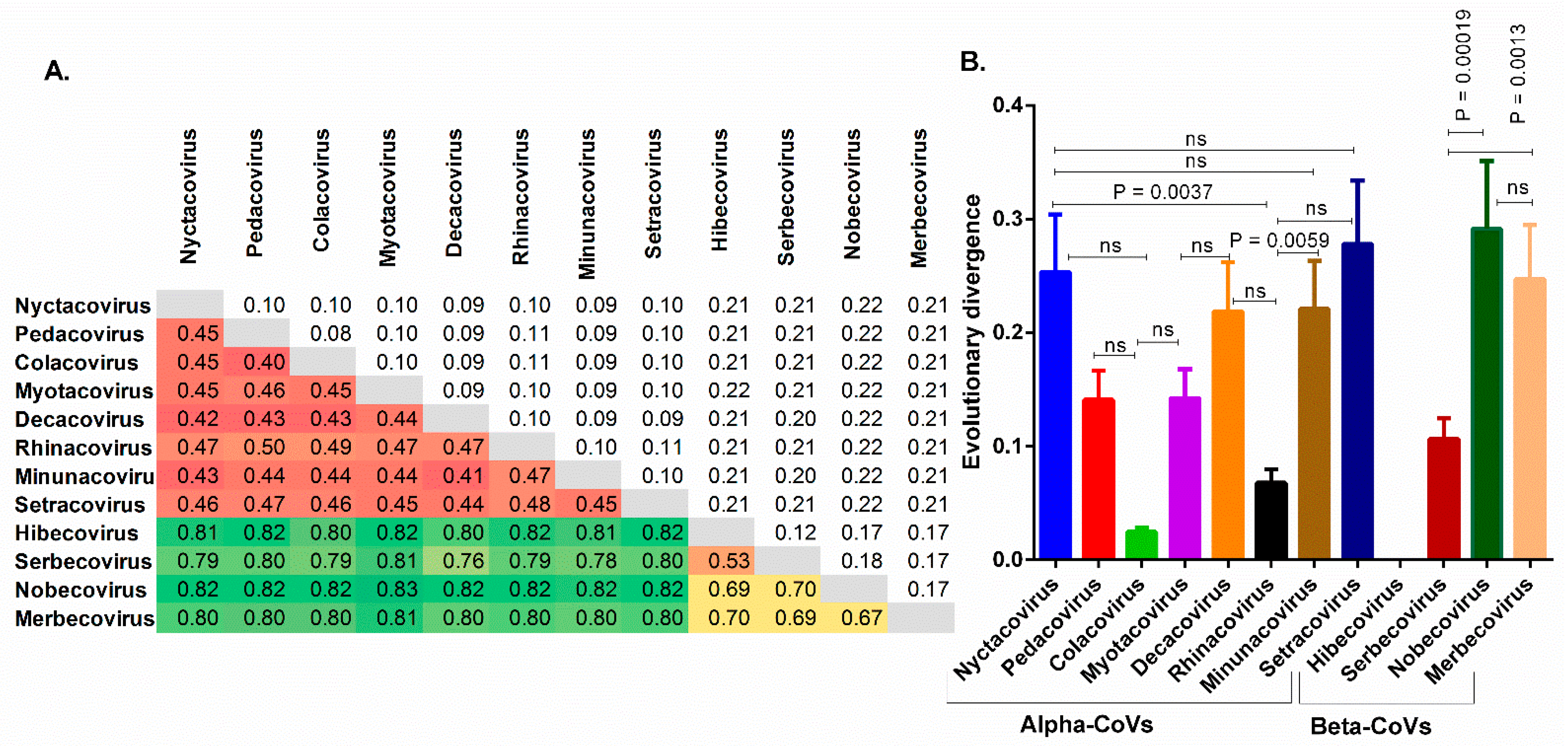
3.1. The Chiroptera-Hosted Coronaviruses (CoVs) Have a Huge Evolutionary Diversity
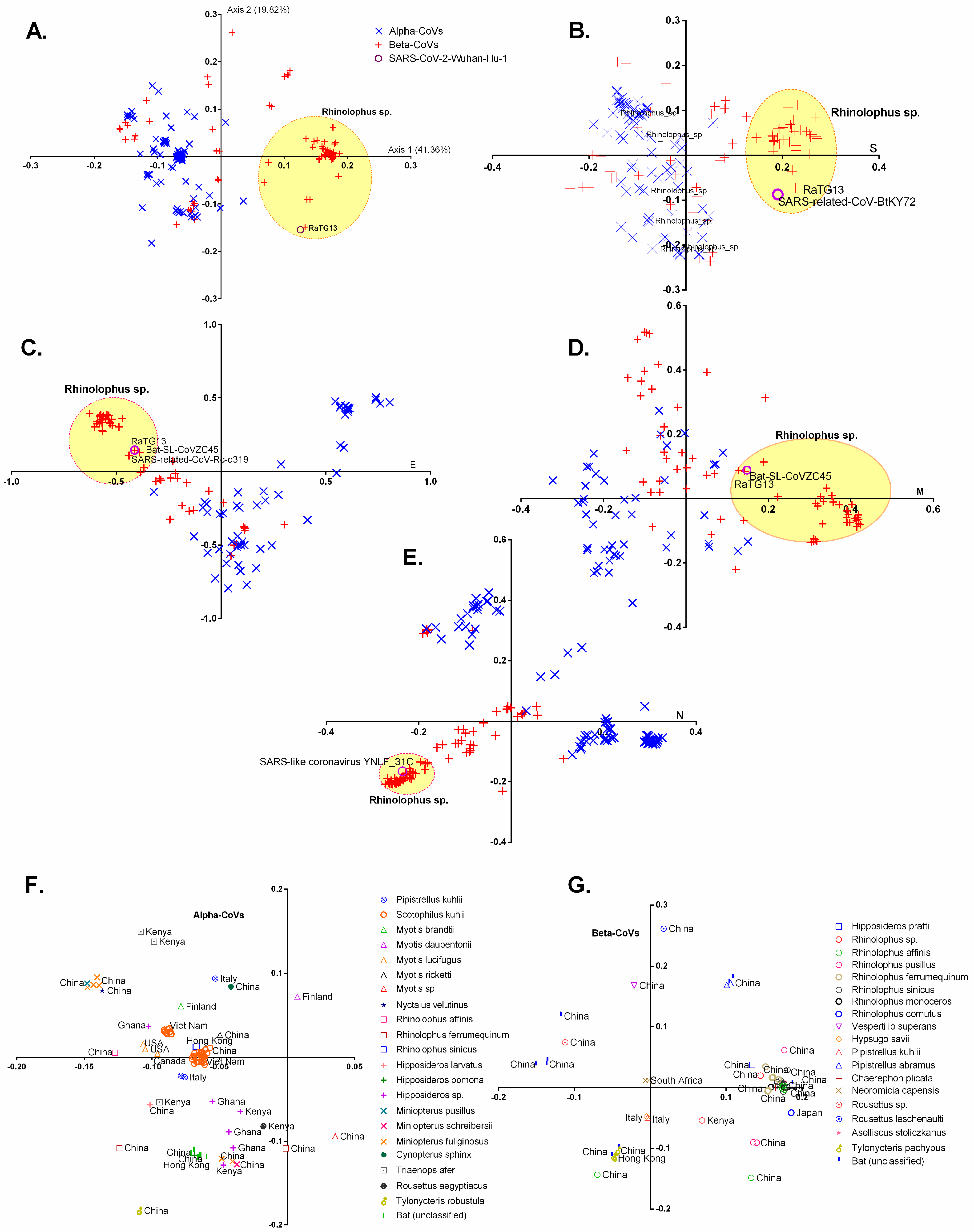
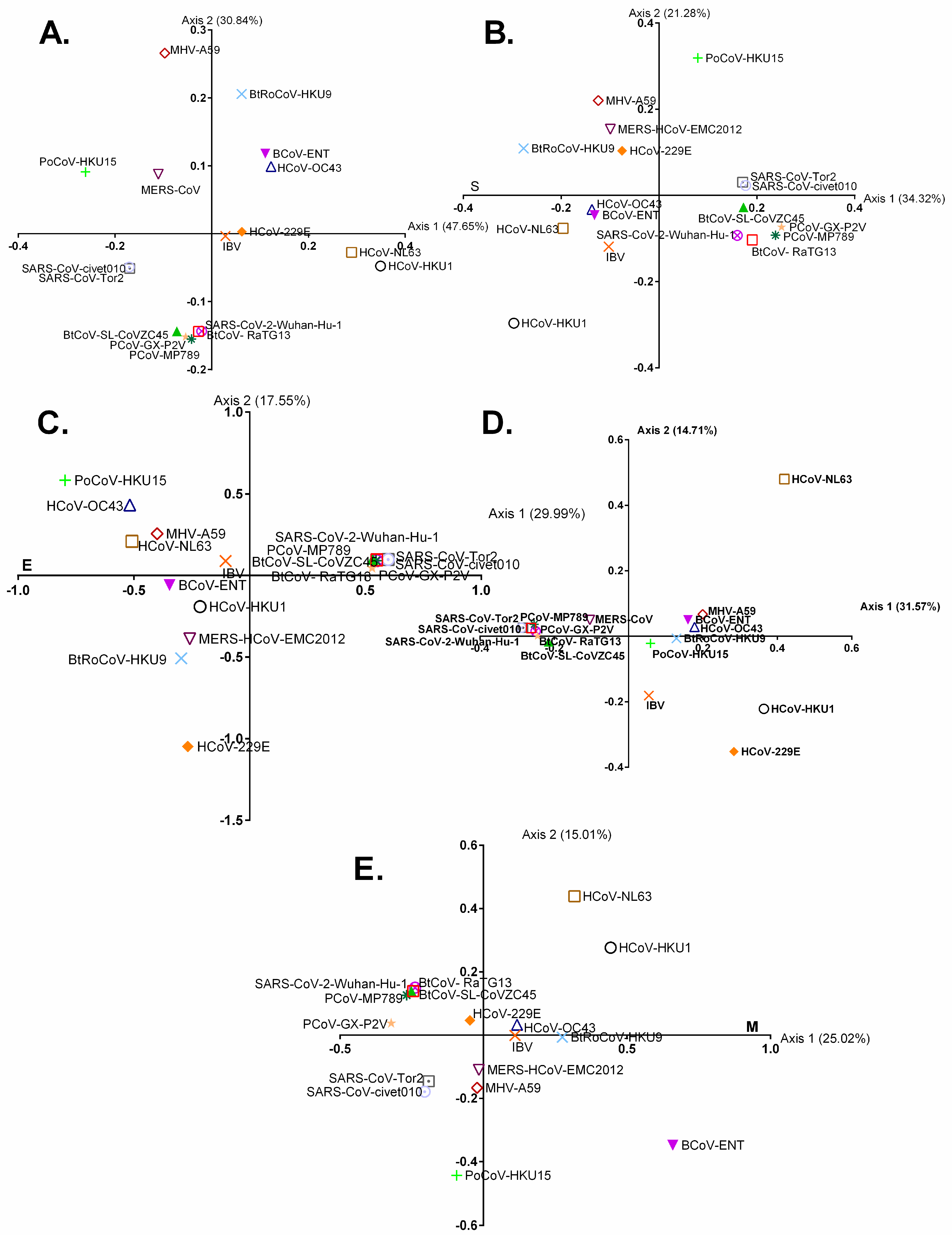
3.2. Geographically Distinct Host Species and Temporo-Spatial Distribution Cooperatively Drive Trends in Codon Usage across Chiroptera-Hosted CoVs
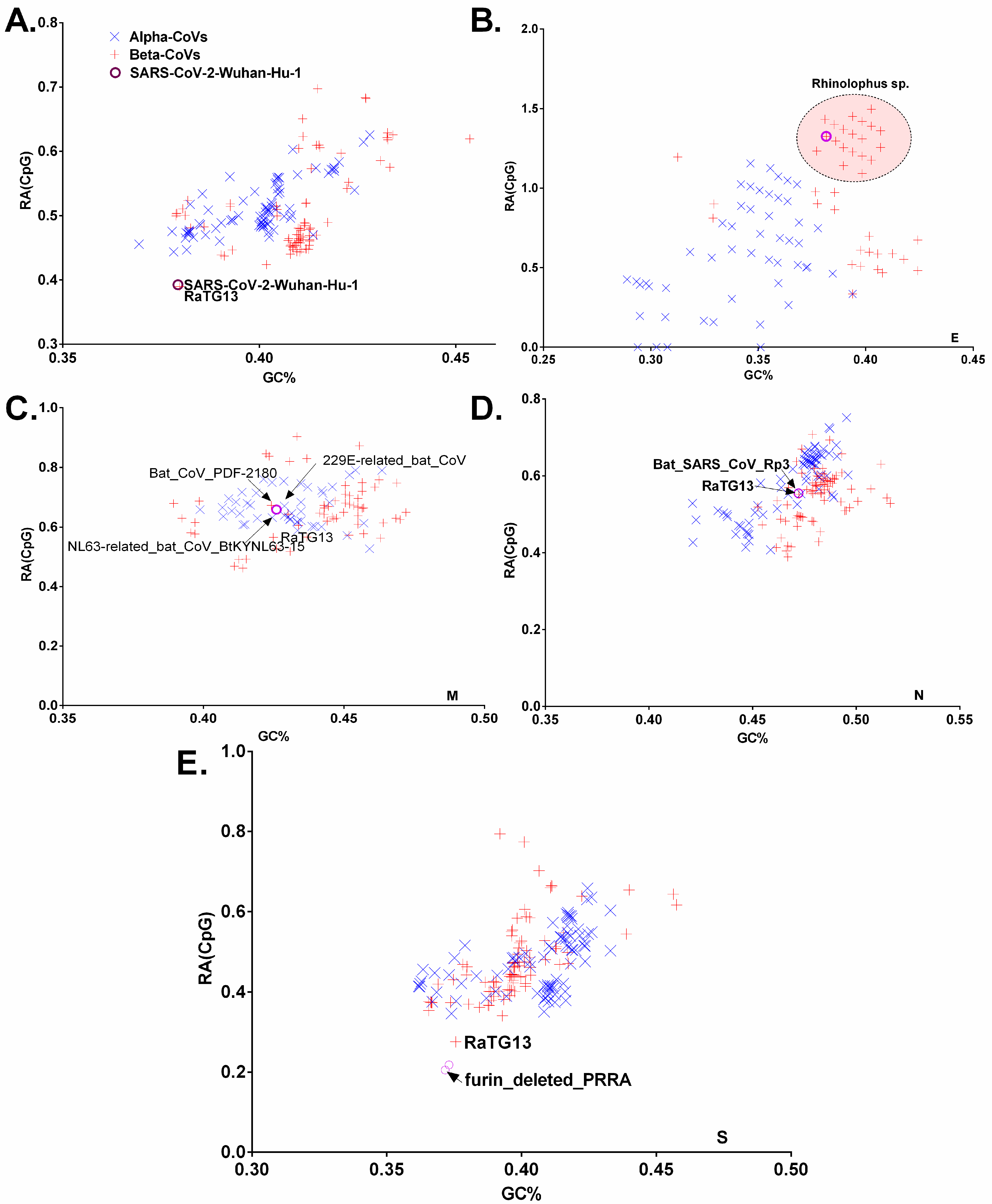
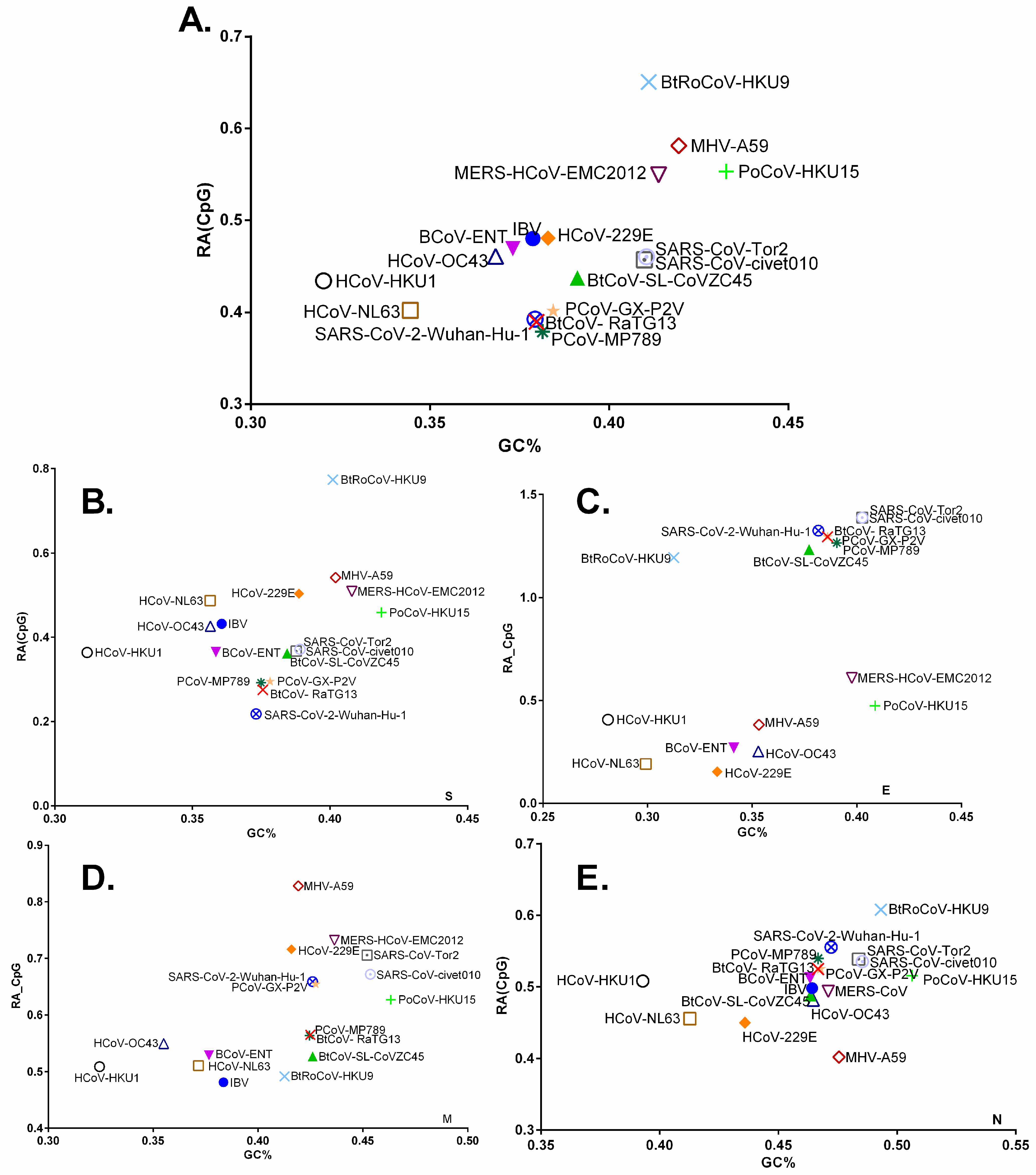
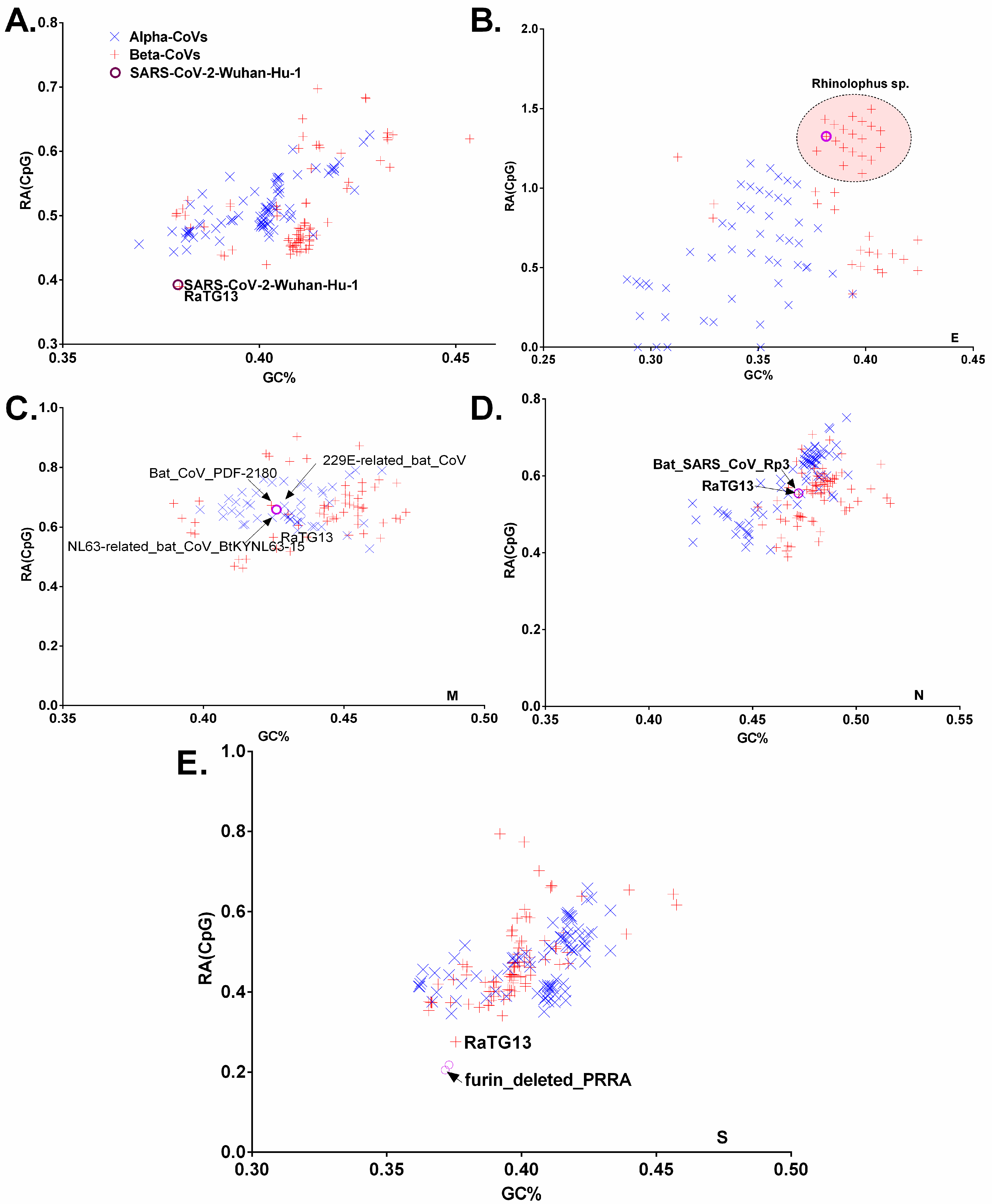
3.3. Chiroptera-Hosted CoVs Were Identified as the Ancestor of SARS-CoV-2 in a Plot of Viral Genomic Relative Abundance of CpG (RACpG) against GC%
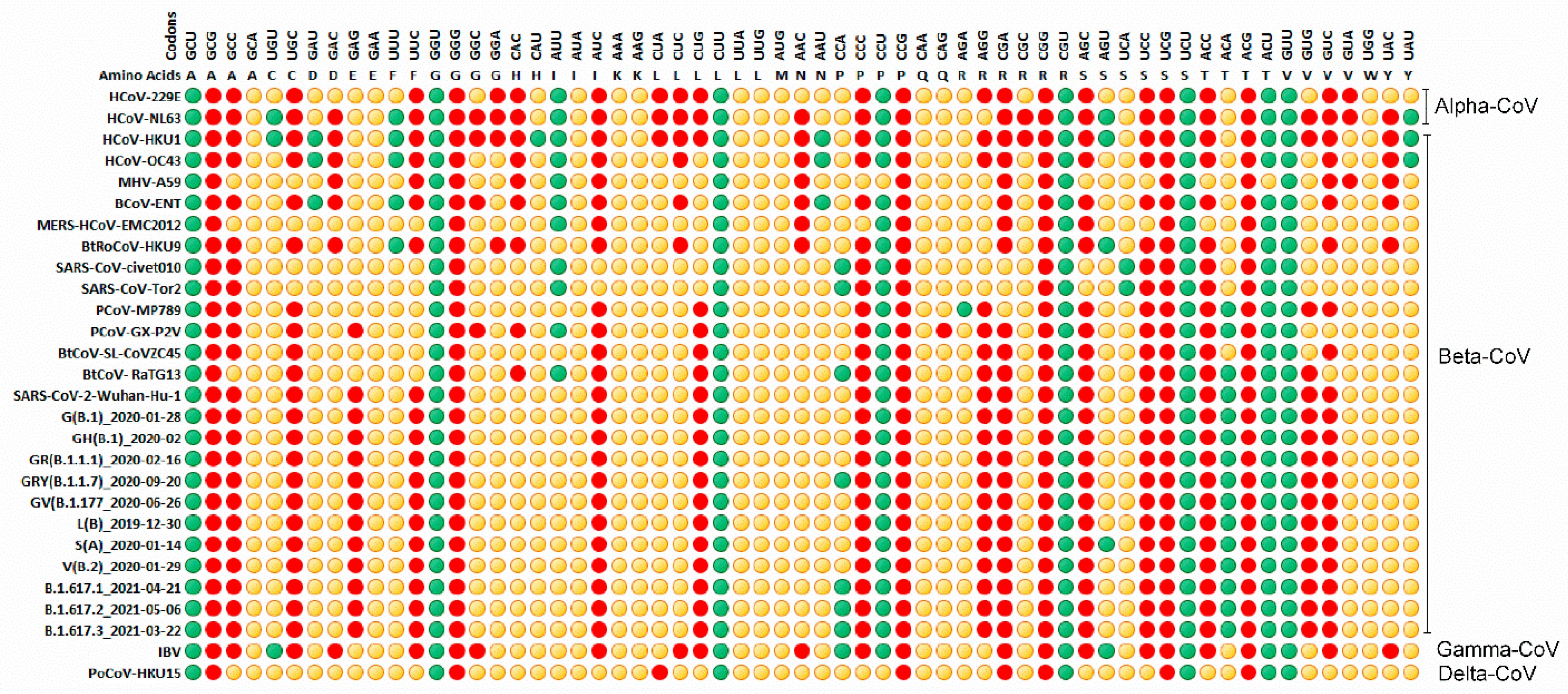
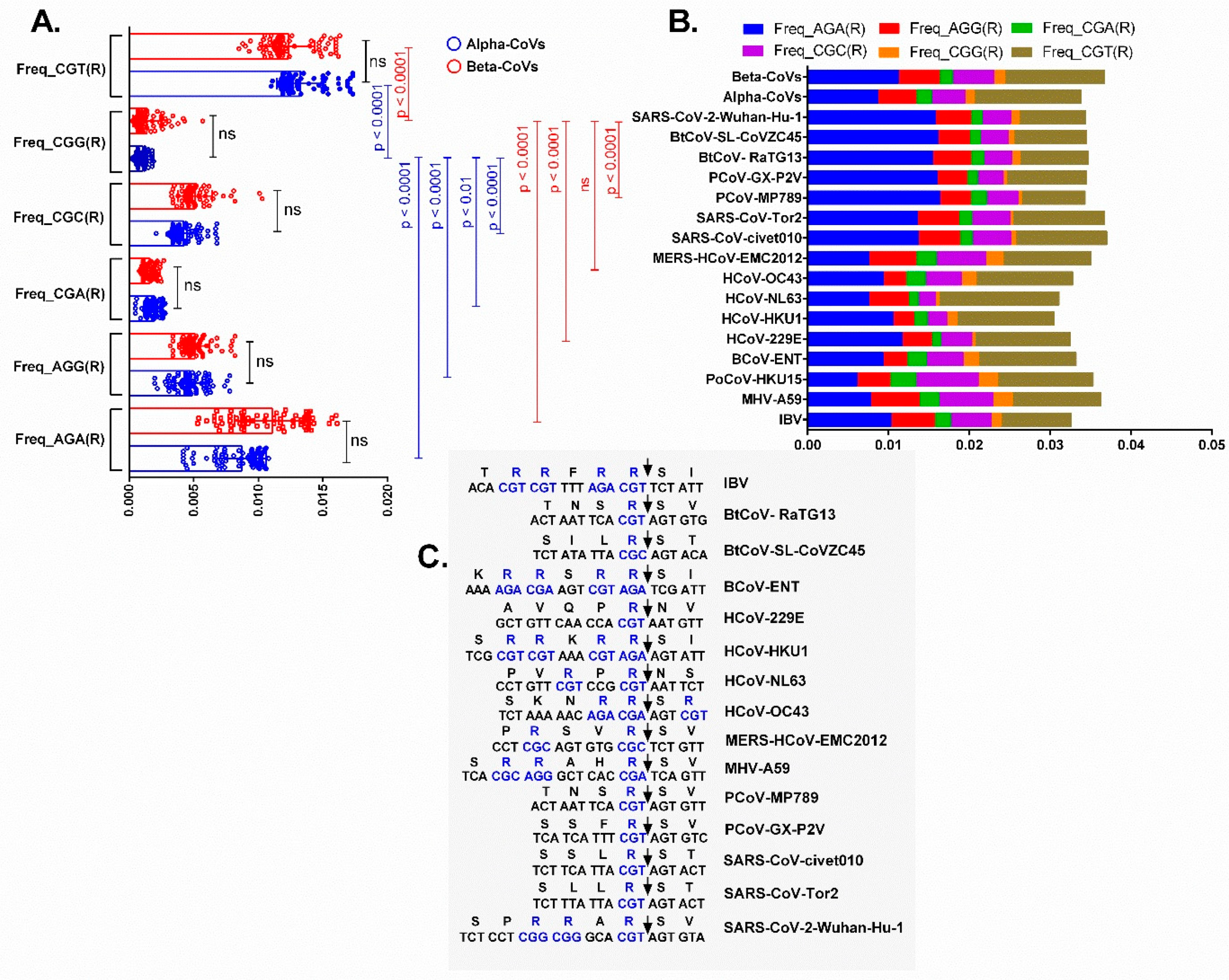
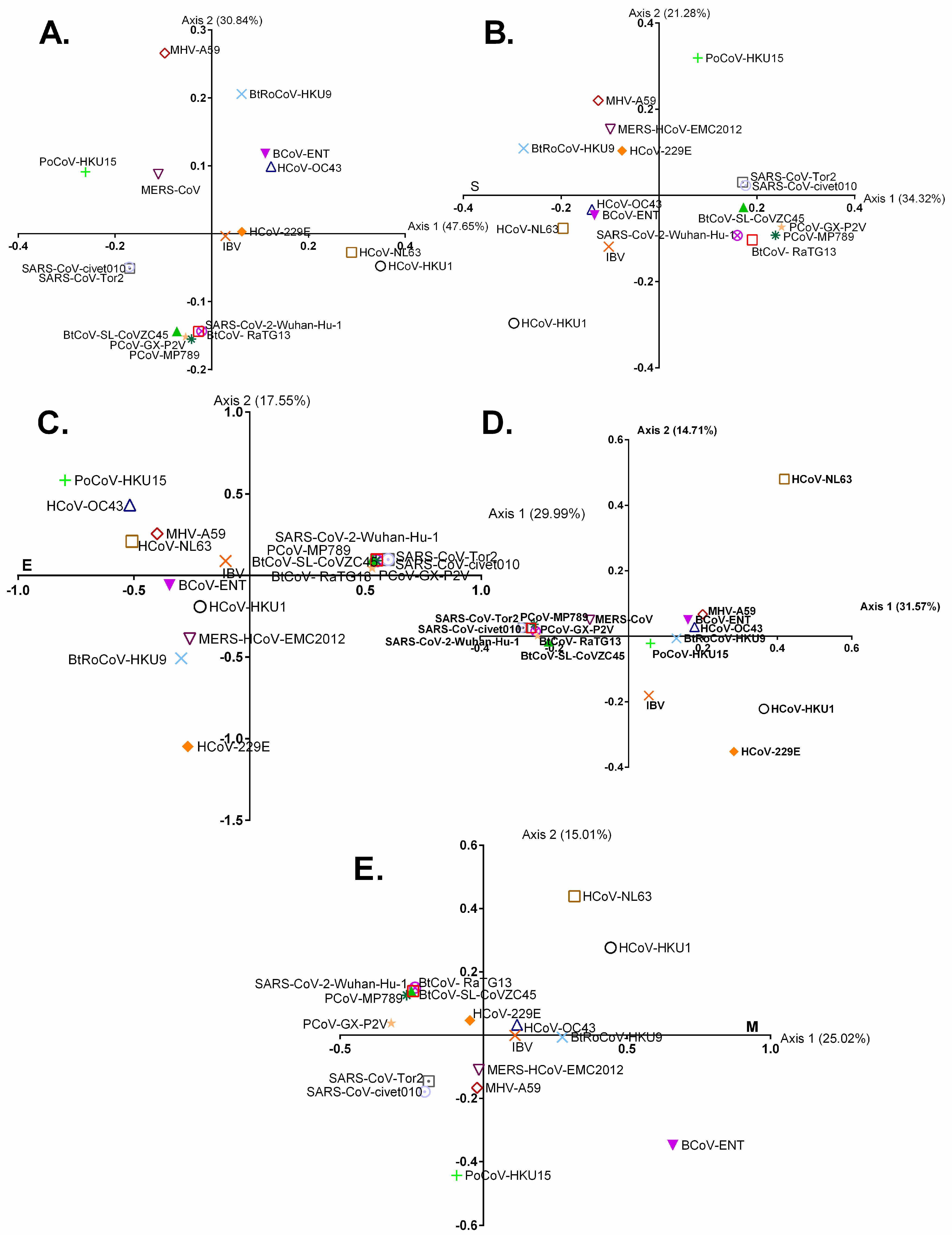
3.4. Relative Synonymous Codon Usage (RSCU) Analysis
3.5. Dissection of the Furin Cleavage Site in CoVs Infecting Several Hosts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Calisher, C.H.; Childs, J.E.; Field, H.E.; Holmes, K.V.; Schountz, T. Bats: Important reservoir hosts of emerging viruses. Clin. Microbiol. Rev. 2006, 19, 531–545. [Google Scholar] [CrossRef] [Green Version]
- Letko, M.; Seifert, S.N.; Olival, K.J.; Plowright, R.K.; Munster, V.J. Bat-borne virus diversity, spillover and emergence. Nat. Rev. Microbiol. 2020, 18, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Simmons, N.B. Order Chiroptera. In Mammal Species of the World: A Taxonomic and Geographic Reference; Wilson, D.E., Reeder, D.M., Eds.; Johns Hopkins University Press: Baltimore, MD, USA, 2005; Volume 2, pp. 312–529. [Google Scholar]
- Tsagkogeorga, G.; Parker, J.; Stupka, E.; Cotton, J.A.; Rossiter, S.J. Phylogenomic analyses elucidate the evolutionary relationships of bats. Curr. Biol. 2013, 23, 2262–2267. [Google Scholar] [CrossRef] [Green Version]
- Amaya, M.; Broder, C.C. Vaccines to Emerging Viruses: Nipah and Hendra. Annu Rev. Virol. 2020, 7, 447–473. [Google Scholar] [CrossRef] [PubMed]
- de Groot, R.J.; Baker, S.C.; Baric, R.S.; Brown, C.S.; Drosten, C.; Enjuanes, L.; Fouchier, R.A.; Galiano, M.; Gorbalenya, A.E.; Memish, Z.A.; et al. Middle East respiratory syndrome coronavirus (MERS-CoV): Announcement of the Coronavirus Study Group. J. Virol. 2013, 87, 7790–7792. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.; Lau, S.K.; Lam, C.S.; Lau, C.C.; Tsang, A.K.; Lau, J.H.; Bai, R.; Teng, J.L.; Tsang, C.C.; Wang, M.; et al. Discovery of seven novel Mammalian and avian coronaviruses in the genus deltacoronavirus supports bat coronaviruses as the gene source of alphacoronavirus and betacoronavirus and avian coronaviruses as the gene source of gammacoronavirus and deltacoronavirus. J. Virol. 2012, 86, 3995–4008. [Google Scholar] [CrossRef] [Green Version]
- ICTV. ICTV Taxonomy History: Cornidovirineae. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 14 July 2021).
- Wong, A.C.P.; Li, X.; Lau, S.K.P.; Woo, P.C.Y. Global Epidemiology of Bat Coronaviruses. Viruses 2019, 11, 174. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decaro, N.; Mari, V.; Campolo, M.; Lorusso, A.; Camero, M.; Elia, G.; Martella, V.; Cordioli, P.; Enjuanes, L.; Buonavoglia, C. Recombinant canine coronaviruses related to transmissible gastroenteritis virus of Swine are circulating in dogs. J. Virol. 2009, 83, 1532–1537. [Google Scholar] [CrossRef] [Green Version]
- Graham, R.L.; Baric, R.S. Recombination, reservoirs, and the modular spike: Mechanisms of coronavirus cross-species transmission. J. Virol. 2010, 84, 3134–3146. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.; Li, K.S.; Huang, Y.; Shek, C.T.; Tse, H.; Wang, M.; Choi, G.K.; Xu, H.; Lam, C.S.; Guo, R.; et al. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol. 2010, 84, 2808–2819. [Google Scholar] [CrossRef] [Green Version]
- Tian, P.F.; Jin, Y.L.; Xing, G.; Qv, L.L.; Huang, Y.W.; Zhou, J.Y. Evidence of recombinant strains of porcine epidemic diarrhea virus, United States, 2013. Emerg. Infect. Dis. 2014, 20, 1735–1738. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Bera, B.C.; Greenbaum, B.D.; Bhatia, S.; Sood, R.; Selvaraj, P.; Anand, T.; Tripathi, B.N.; Virmani, N. Revelation of Influencing Factors in Overall Codon Usage Bias of Equine Influenza Viruses. PLoS ONE 2016, 11, e0154376. [Google Scholar] [CrossRef] [Green Version]
- Kumar, N.; Kulkarni, D.D.; Lee, B.; Kaushik, R.; Bhatia, S.; Sood, R.; Pateriya, A.K.; Bhat, S.; Singh, V.P. Evolution of Codon Usage Bias in Henipaviruses Is Governed by Natural Selection and Is Host-Specific. Viruses 2018, 10, 604. [Google Scholar] [CrossRef] [Green Version]
- Xia, X. Extreme Genomic CpG Deficiency in SARS-CoV-2 and Evasion of Host Antiviral Defense. Mol. Biol. Evol. 2020, 37, 2699–2705. [Google Scholar] [CrossRef]
- Kumar, N.; Kaushik, R.; Tennakoon, C.; Uversky, V.N.; Longhi, S.; Zhang, K.Y.J.; Bhatia, S. Insights into the evolutionary forces that shape the codon usage in the viral genome segments encoding intrinsically disordered protein regions. Brief. Bioinform. 2021, 22, bbab145. [Google Scholar] [CrossRef] [PubMed]
- Mordstein, C.; Cano, L.; Morales, A.C.; Young, B.; Ho, A.T.; Rice, A.M.; Liss, M.; Hurst, L.D.; Kudla, G. Transcription, mRNA Export, and Immune Evasion Shape the Codon Usage of Viruses. Genome Biol. Evol. 2021, 13, evab106. [Google Scholar] [CrossRef]
- Pickett, B.E.; Sadat, E.L.; Zhang, Y.; Noronha, J.M.; Squires, R.B.; Hunt, V.; Liu, M.; Kumar, S.; Zaremba, S.; Gu, Z.; et al. ViPR: An open bioinformatics database and analysis resource for virology research. Nucleic Acids Res. 2012, 40, D593–D598. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Nei, M.; Kumar, S. Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. USA 2004, 101, 11030–11035. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, P. SSE: A nucleotide and amino acid sequence analysis platform. BMC Res. Notes 2012, 5, 50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, X. DAMBE7: New and Improved Tools for Data Analysis in Molecular Biology and Evolution. Mol. Biol. Evol. 2018, 35, 1550–1552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karlin, S.; Doerfler, W.; Cardon, L.R. Why is CpG suppressed in the genomes of virtually all small eukaryotic viruses but not in those of large eukaryotic viruses? J. Virol. 1994, 68, 2889–2897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharp, P.M.; Li, W.H. Codon usage in regulatory genes in Escherichia coli does not reflect selection for ‘rare’ codons. Nucleic Acids Res. 1986, 14, 7737–7749. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, E.H.; Smith, D.K.; Rabadan, R.; Peiris, M.; Poon, L.L. Codon usage bias and the evolution of influenza A viruses. Codon Usage Biases of Influenza Virus. BMC Evol. Biol. 2010, 10, 253. [Google Scholar] [CrossRef] [Green Version]
- Greenacre, M.J. Theory and Applications of Correspondence Analysis; Academic Press: London, UK, 1984. [Google Scholar]
- Jacob Machado, D.; Scott, R.; Guirales, S.; Janies, D.A. Fundamental evolution of all Orthocoronavirinae including three deadly lineages descendent from Chiroptera-hosted coronaviruses: SARS-CoV, MERS-CoV and SARS-CoV-2. Cladistics 2021. [Google Scholar] [CrossRef]
- Vijaykrishna, D.; Smith, G.J.; Zhang, J.X.; Peiris, J.S.; Chen, H.; Guan, Y. Evolutionary insights into the ecology of coronaviruses. J. Virol. 2007, 81, 4012–4020. [Google Scholar] [CrossRef] [Green Version]
- Woo, P.C.; Lau, S.K.; Huang, Y.; Yuen, K.Y. Coronavirus diversity, phylogeny and interspecies jumping. Exp. Biol. Med. (Maywood) 2009, 234, 1117–1127. [Google Scholar] [CrossRef] [Green Version]
- Lau, S.K.; Woo, P.C.; Li, K.S.; Huang, Y.; Tsoi, H.W.; Wong, B.H.; Wong, S.S.; Leung, S.Y.; Chan, K.H.; Yuen, K.Y. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. USA 2005, 102, 14040–14045. [Google Scholar] [CrossRef] [Green Version]
- Subudhi, S.; Rapin, N.; Bollinger, T.K.; Hill, J.E.; Donaldson, M.E.; Davy, C.M.; Warnecke, L.; Turner, J.M.; Kyle, C.J.; Willis, C.K.R.; et al. A persistently infecting coronavirus in hibernating Myotis lucifugus, the North American little brown bat. J. Gen. Virol. 2017, 98, 2297–2309. [Google Scholar] [CrossRef]
- Watanabe, S.; Masangkay, J.S.; Nagata, N.; Morikawa, S.; Mizutani, T.; Fukushi, S.; Alviola, P.; Omatsu, T.; Ueda, N.; Iha, K.; et al. Bat coronaviruses and experimental infection of bats, the Philippines. Emerg. Infect. Dis. 2010, 16, 1217–1223. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Shi, Z.; Yu, M.; Ren, W.; Smith, C.; Epstein, J.H.; Wang, H.; Crameri, G.; Hu, Z.; Zhang, H.; et al. Bats are natural reservoirs of SARS-like coronaviruses. Science 2005, 310, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Tian, X.; Qin, P.; Wang, B.; Zhao, P.; Yang, Y.L.; Wang, L.; Wang, D.; Song, Y.; Zhang, X.; et al. Discovery of a novel swine enteric alphacoronavirus (SeACoV) in southern China. Vet. Microbiol. 2017, 211, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Fan, H.; Lan, T.; Yang, X.L.; Shi, W.F.; Zhang, W.; Zhu, Y.; Zhang, Y.W.; Xie, Q.M.; Mani, S.; et al. Fatal swine acute diarrhoea syndrome caused by an HKU2-related coronavirus of bat origin. Nature 2018, 556, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Lau, S.K.; Feng, Y.; Chen, H.; Luk, H.K.; Yang, W.H.; Li, K.S.; Zhang, Y.Z.; Huang, Y.; Song, Z.Z.; Chow, W.N.; et al. Severe Acute Respiratory Syndrome (SARS) Coronavirus ORF8 Protein Is Acquired from SARS-Related Coronavirus from Greater Horseshoe Bats through Recombination. J. Virol. 2015, 89, 10532–10547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Z.; Yang, L.; Ren, X.; Zhang, J.; Yang, F.; Zhang, S.; Jin, Q. ORF8-Related Genetic Evidence for Chinese Horseshoe Bats as the Source of Human Severe Acute Respiratory Syndrome Coronavirus. J. Infect. Dis. 2016, 213, 579–583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meagher, J.L.; Takata, M.; Gonçalves-Carneiro, D.; Keane, S.C.; Rebendenne, A.; Ong, H.; Orr, V.K.; MacDonald, M.R.; Stuckey, J.A.; Bieniasz, P.D.; et al. Structure of the zinc-finger antiviral protein in complex with RNA reveals a mechanism for selective targeting of CG-rich viral sequences. Proc. Natl. Acad. Sci. USA 2019, 116, 24303–24309. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Patnaik, S.K.; Taggart, R.T.; Baysal, B.E. The double-domain cytidine deaminase APOBEC3G is a cellular site-specific RNA editing enzyme. Sci. Rep. 2016, 6, 39100. [Google Scholar] [CrossRef]
- Sharma, S.; Patnaik, S.K.; Taggart, R.T.; Kannisto, E.D.; Enriquez, S.M.; Gollnick, P.; Baysal, B.E. APOBEC3A cytidine deaminase induces RNA editing in monocytes and macrophages. Nat. Commun 2015, 6, 6881. [Google Scholar] [CrossRef]
- Sharma, S.; Wang, J.; Alqassim, E.; Portwood, S.; Cortes Gomez, E.; Maguire, O.; Basse, P.H.; Wang, E.S.; Segal, B.H.; Baysal, B.E. Mitochondrial hypoxic stress induces widespread RNA editing by APOBEC3G in natural killer cells. Genome Biol. 2019, 20, 37. [Google Scholar] [CrossRef] [Green Version]
- Digard, P.; Lee, H.M.; Sharp, C.; Grey, F.; Gaunt, E. Intra-genome variability in the dinucleotide composition of SARS-CoV-2. Virus Evol. 2020, 6, veaa057. [Google Scholar] [CrossRef] [PubMed]
- Johnson, B.A.; Xie, X.; Kalveram, B.; Lokugamage, K.G.; Muruato, A.; Zou, J.; Zhang, X.; Juelich, T.; Smith, J.K.; Zhang, L.; et al. Furin Cleavage Site Is Key to SARS-CoV-2 Pathogenesis. bioRxiv 2020. [Google Scholar] [CrossRef]
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol 2021, 6, 899–909. [Google Scholar] [CrossRef]
- Zhou, H.; Chen, X.; Hu, T.; Li, J.; Song, H.; Liu, Y.; Wang, P.; Liu, D.; Yang, J.; Holmes, E.C.; et al. A Novel Bat Coronavirus Closely Related to SARS-CoV-2 Contains Natural Insertions at the S1/S2 Cleavage Site of the Spike Protein. Curr. Biol. 2020, 30, 2196–2203. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, N.; Kaushik, R.; Tennakoon, C.; Uversky, V.N.; Mishra, A.; Sood, R.; Srivastava, P.; Tripathi, M.; Zhang, K.Y.J.; Bhatia, S. Evolutionary Signatures Governing the Codon Usage Bias in Coronaviruses and Their Implications for Viruses Infecting Various Bat Species. Viruses 2021, 13, 1847. https://doi.org/10.3390/v13091847
Kumar N, Kaushik R, Tennakoon C, Uversky VN, Mishra A, Sood R, Srivastava P, Tripathi M, Zhang KYJ, Bhatia S. Evolutionary Signatures Governing the Codon Usage Bias in Coronaviruses and Their Implications for Viruses Infecting Various Bat Species. Viruses. 2021; 13(9):1847. https://doi.org/10.3390/v13091847
Chicago/Turabian StyleKumar, Naveen, Rahul Kaushik, Chandana Tennakoon, Vladimir N. Uversky, Anamika Mishra, Richa Sood, Pratiksha Srivastava, Meghna Tripathi, Kam Y. J. Zhang, and Sandeep Bhatia. 2021. "Evolutionary Signatures Governing the Codon Usage Bias in Coronaviruses and Their Implications for Viruses Infecting Various Bat Species" Viruses 13, no. 9: 1847. https://doi.org/10.3390/v13091847
APA StyleKumar, N., Kaushik, R., Tennakoon, C., Uversky, V. N., Mishra, A., Sood, R., Srivastava, P., Tripathi, M., Zhang, K. Y. J., & Bhatia, S. (2021). Evolutionary Signatures Governing the Codon Usage Bias in Coronaviruses and Their Implications for Viruses Infecting Various Bat Species. Viruses, 13(9), 1847. https://doi.org/10.3390/v13091847