
Phylogenomic Characterization of Lopma Virus and Praja Virus, Two Novel Rodent-Borne Arteriviruses

 , ,
, ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Illumina Sequencing
2.2. Genome Assembly
2.3. Dataset Compilation and Phylogenetic Analysis
2.4. Co-Divergence Analysis
3. Results
3.1. Detection of Praja and Lopma Viruses
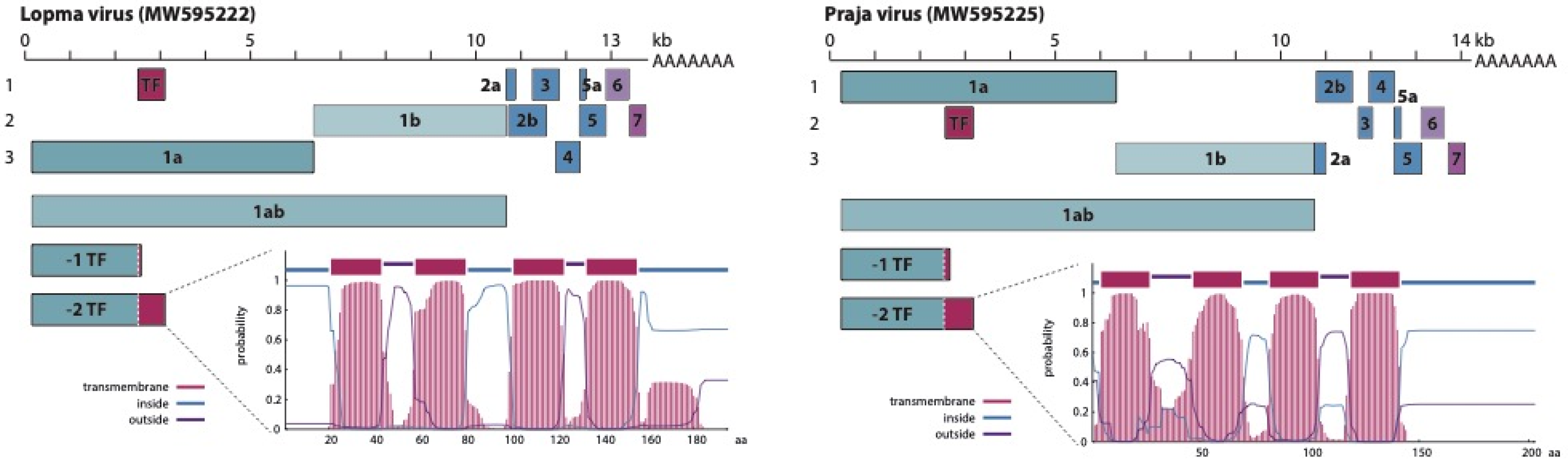
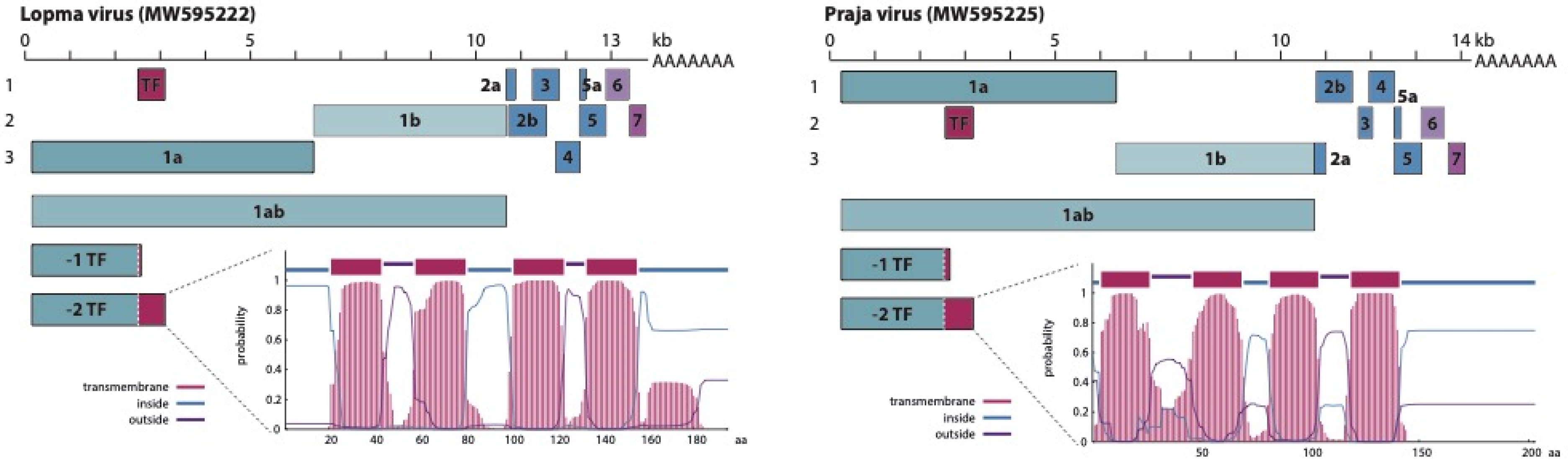
3.2. Genome Organization
3.3. Phylogenetic Analysis of the Mammalian-Borne Arteriviruses
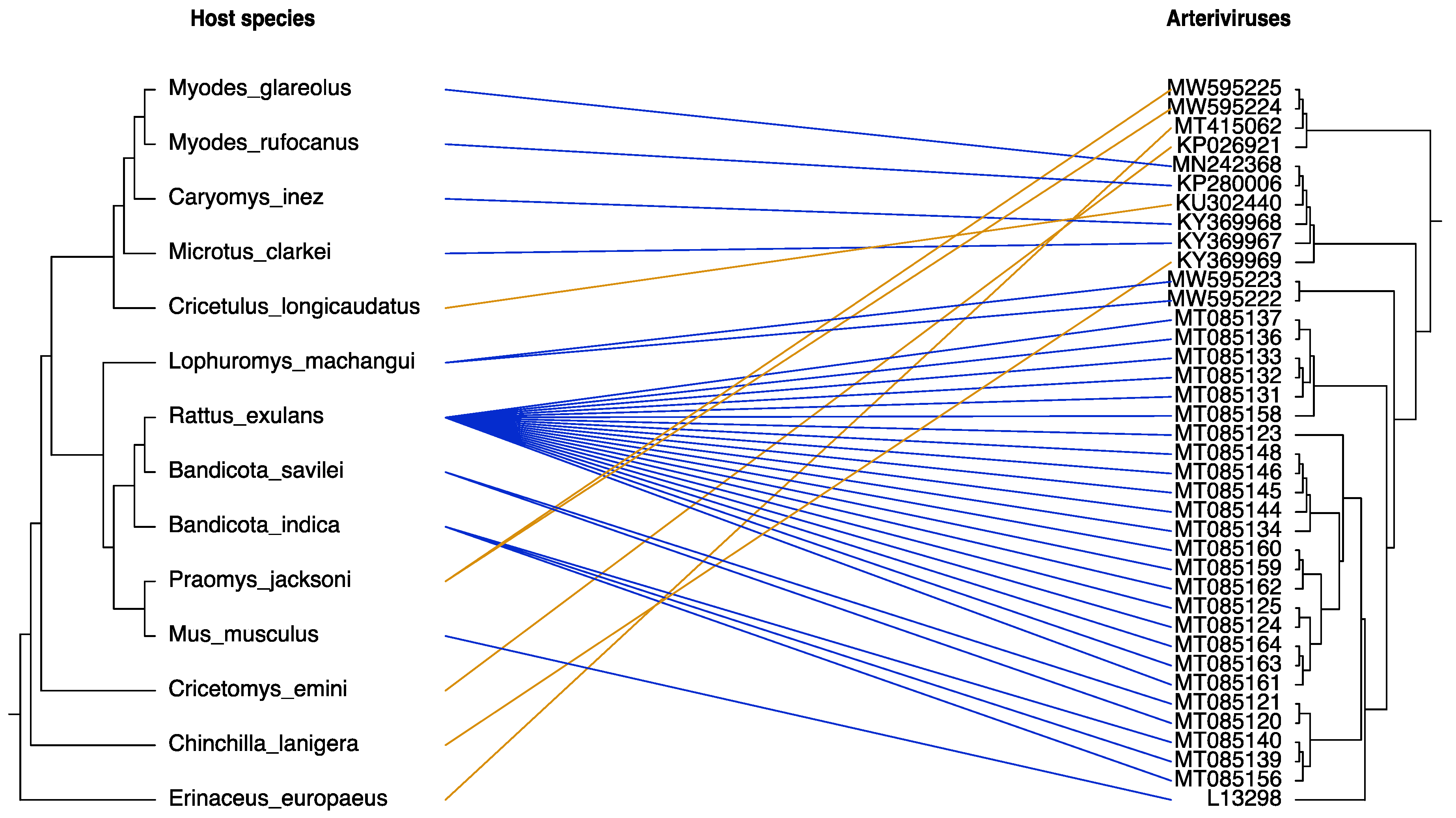
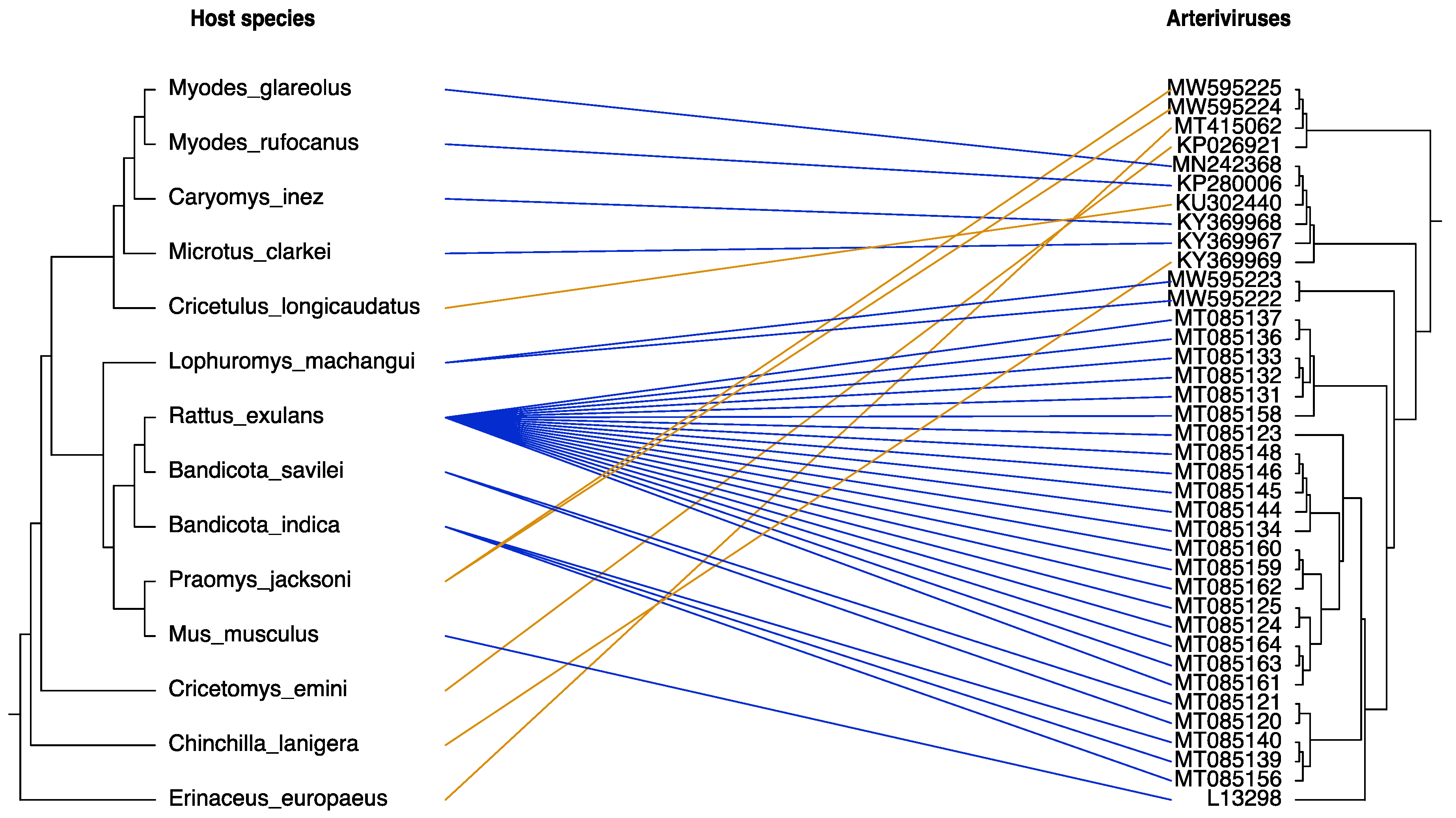
3.4. Co-Divergence of Rodent-Borne Arteriviruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gortazar, C.; Reperant, L.A.; Kuiken, T.; de la Fuente, J.; Boadella, M.; Martínez-Lopez, B.; Ruiz-Fons, F.; Estrada-Peña, A.; Drosten, C.; Medley, G.; et al. Crossing the Interspecies Barrier: Opening the Door to Zoonotic Pathogens. PLoS Pathog. 2014, 10, e1004129. [Google Scholar] [CrossRef] [Green Version]
- Lipkin, W.I.; Anthony, S.J. Virus hunting. Virology 2015, 479–480, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Mollentze, N.; Streicker, D.G. Viral zoonotic risk is homogenous among taxonomic orders of mammalian and avian reservoir hosts. Proc. Natl. Acad. Sci. USA 2020, 117, 9423–9430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luis, A.D.; Hayman, D.T.S.; O’Shea, T.J.; Cryan, P.M.; Gilbert, A.T.; Pulliam, J.R.C.; Mills, J.N.; Timonin, M.E.; Willis, C.K.R.; Cunningham, A.A.; et al. A comparison of bats and rodents as reservoirs of zoonotic viruses: Are bats special? Proc. Biol. Sci. 2013, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bailey, A.L.; Lauck, M.; Sibley, S.D.; Friedrich, T.; Kuhn, J.H.; Freimer, N.B.; Jasinska, A.J.; Phillips-Conroy, J.E.; Jolly, C.J.; Marx, P.A.; et al. Zoonotic Potential of Simian Arteriviruses. J. Virol. 2016, 90, 630–635. [Google Scholar] [CrossRef] [Green Version]
- Balasuriya, U.B.; MacLachlan, N.J. The immune response to equine arteritis virus: Potential lessons for other arteriviruses. Veter.-Immunol. Immunopathol. 2004, 102, 107–129. [Google Scholar] [CrossRef]
- Lunney, J.K.; Benfield, D.A.; Rowland, R.R. Porcine reproductive and respiratory syndrome virus: An update on an emerging and re-emerging viral disease of swine. Virus Res. 2010, 154, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Giles, J.; Perrott, M.; Roe, W.; Dunowska, M. The aetiology of wobbly possum disease: Reproduction of the disease with purified nidovirus. Virology 2016, 491, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Mackintosh, C.G.; Crawford, J.L.; Thompson, E.G.; McLeod, B.J.; Gill, J.M.; O’Keefe, J.S. A newly discovered disease of the brushtail possum: Wobbly possum syndrome. N. Z. Vet. J. 1995, 43, 126. [Google Scholar] [CrossRef] [PubMed]
- Rowson, K.E.; Mahy, B.W. Lactate dehydrogenase-elevating virus. J. Gen. Virol. 1985, 66 Pt 11, 2297–2312. [Google Scholar] [CrossRef]
- Wahl-Jensen, V.; Johnson, J.C.; Lauck, M.; Weinfurter, J.T.; Moncla, L.H.; Weiler, A.M.; Charlier, O.; Rojas, O.; Byrum, R.; Ragland, D.R.; et al. Divergent Simian Arteriviruses Cause Simian Hemorrhagic Fever of Differing Severities in Macaques. mBio 2016, 7, e02009–e02015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijder, E.J.; Kikkert, M.; Fang, Y. Arterivirus molecular biology and pathogenesis. J. Gen. Virol. 2013, 94, 2141–2163. [Google Scholar] [CrossRef] [PubMed]
- Posthuma, C.C.; Velthuis, A.T.; Snijder, E. Nidovirus RNA polymerases: Complex enzymes handling exceptional RNA genomes. Virus Res. 2017, 234, 58–73. [Google Scholar] [CrossRef] [PubMed]
- Godeny, E.K.; de Vries, A.A.; Wang, X.C.; Smith, S.L.; de Groot, R.J. Identification of the leader-body junctions for the viral subgenomic mRNAs and organization of the simian hemorrhagic fever virus genome: Evidence for gene duplication during arterivirus evolution. J. Virol. 1998, 72, 862–867. [Google Scholar] [CrossRef] [Green Version]
- Vatter, H.A.; Di, H.; Donaldson, E.F.; Baric, R.S.; Brinton, M.A. Each of the eight simian hemorrhagic fever virus minor structural proteins is functionally important. Virology 2014, 462–463, 351–362. [Google Scholar] [CrossRef] [Green Version]
- Di, H.; Madden, J.C., Jr.; Morantz, E.K.; Tang, H.Y.; Graham, R.L.; Baric, R.S.; Brinton, M.A. Expanded subgenomic mRNA transcriptome and coding capacity of a nidovirus. Proc. Natl. Acad. Sci. USA 2017, 114, E8895–E8904. [Google Scholar] [CrossRef] [Green Version]
- ICTV. ICTV 2019 Master Species List (MSL35). 2020. Available online: https://talk.ictvonline.org/files/master-species-lists/m/msl/9601 (accessed on 3 December 2020).
- Kesäniemi, J.; Lavrinienko, A.; Tukalenko, E.; Mappes, T.; Watts, P.C.; Jurvansuu, J. Infection Load and Prevalence of Novel Viruses Identified from the Bank Vole Do Not Associate with Exposure to Environmental Radioactivity. Viruses 2019, 12, 44. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Lin, X.-D.; Chen, X.; Tian, J.-H.; Chen, L.-J.; Li, K.; Wang, W.; Eden, J.-S.; Shen, J.-J.; Liu, L.; et al. The evolutionary history of vertebrate RNA viruses. Nature 2018, 556, 197–202. [Google Scholar] [CrossRef]
- Wu, Z.; Han, Y.; Liu, B.; Li, H.; Zhu, G.; Latinne, A.; Dong, J.; Sun, L.; Du, J.; Zhou, S.; et al. Decoding the RNA Viromes of Rodent Lungs Provides New Visions into the Origin and Evolution Pattern of Rodent-Borne Diseases in Mainland Southeast Asia. PREPRINT (Version 1) Available at Research Square. 2020. Available online: https://www.researchsquare.com/article/rs-17323/v1 (accessed on 22 December 2020).
- Van de Perre, F.; Willig, M.R.; Presley, S.J.; Bapeamoni Andemwana, F.; Beeckman, H.; Boeckx, P.; Cooleman, S.; de Haan, M.; De Kesel, A.; Dessein, S.; et al. Reconciling biodiversity and carbon stock conservation in an Afrotropical forest landscape. Sci. Adv. 2018, 4, eaar6603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gryseels, S.; Baird, S.J.E.; Borremans, B.; Makundi, R.; Leirs, H.; Gouy de Bellocq, J. When Viruses Don’t Go Viral: The Importance of Host Phylogeographic Structure in the Spatial Spread of Arenaviruses. PLoS Pathog. 2017, 13, e1006073. [Google Scholar] [CrossRef] [Green Version]
- Tesikova, J.; Bryjova, A.; Bryja, J.; Lavrenchenko, L.A.; Gouy de Bellocq, J. Hantavirus Strains in East Africa Related to Western African Hantaviruses. Vector Borne Zoonotic Dis. 2017, 17, 278–280. [Google Scholar] [CrossRef] [PubMed]
- Gryseels, S.; Rieger, T.; Oestereich, L.; Cuypers, B.; Borremans, B.; Makundi, R.; Leirs, H.; Gunther, S.; Gouy de Bellocq, J. Gairo virus, a novel arenavirus of the widespread Mastomys natalensis: Genetically divergent, but ecologically similar to Lassa and Morogoro viruses. Virology 2015, 476, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Makundi, R.H.; Massawe, A.W.; Borremans, B.; Laudisoit, A.; Katakweba, A. We are connected: Flea–host association networks in the plague outbreak focus in the Rift Valley, northern Tanzania. Wildl. Res. 2015, 42, 196–206. [Google Scholar] [CrossRef]
- Massawe, A.W.; Makundi, R.H.; Mulungu, L.S.; Katakweba, A.; Shayo, T.N. Breeding dynamics of rodent species inhabiting farm–fallow mosaic fields in Central Tanzania. Afr. Zool. 2012, 47, 128–137. [Google Scholar] [CrossRef]
- Meheretu, Y.; Čížková, D.; Těšíková, J.; Welegerima, K.; Tomas, Z.; Kidane, D.; Girmay, K.; Schmidt-Chanasit, J.; Bryja, J.; Günther, S.; et al. High Diversity of RNA Viruses in Rodents, Ethiopia. Emerg. Infect. Dis. 2012, 18, 2047–2050. [Google Scholar] [CrossRef] [PubMed]
- Gouy de Bellocq, J.; Borremans, B.; Katakweba, A.; Makundi, R.; Baird, S.J.; Becker-Ziaja, B.; Günther, S.; Leirs, H. Sympatric Occurrence of 3 Arenaviruses, Tanzania. Emerg. Infect. Dis. 2010, 16, 692–695. [Google Scholar] [CrossRef] [PubMed]
- Laudisoit, A.; Leirs, H.; Makundi, R.; Krasnov, B.R. Seasonal and habitat dependence of fleas parasitic on small mammals in Tanzania. Integr. Zool. 2009, 4, 196–212. [Google Scholar] [CrossRef]
- Bletsa, M.; Vrancken, B.; Gryseels, S.; Boonen, I.; Fikatas, A.; Li, Y.; Laudisoit, A.; Lequime, S.; Bryja, J.; Makun-di, R.; et al. Molecular detection and genomic characterisation of diverse hepaciviruses in African rodents. Virus Evol. 2021, 7, veab036. [Google Scholar] [CrossRef] [PubMed]
- National Center for Biotechnology Information (NCBI). Basic Local Alignment Search Tool (BLAST). 2018. Available online: https://blast.ncbi.nlm.nih.gov/blast.cgi (accessed on 15 January 2018).
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30, 3276–3278. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Chetvernin, V.; Tatusova, T. Improvements to pairwise sequence comparison (PASC): A genome-based web tool for virus classification. Arch. Virol. 2014, 159, 3293–3304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dastjerdi, A.; Inglese, N.; Partridge, T.; Karuna, S.; Everest, D.J.; Frossard, J.-P.; Dagleish, M.P.; Stidworthy, M.F. Novel Arterivirus Associated with Outbreak of Fatal Encephalitis in European Hedgehogs, England, 2019. Emerg. Infect. Dis. 2021, 27, 578–581. [Google Scholar] [CrossRef]
- Upham, N.S.; Esselstyn, J.A.; Jetz, W. Inferring the mammal tree: Species-level sets of phylogenies for questions in ecology, evolution, and conservation. PLoS Biol. 2019, 17, e3000494. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Schliep, K. ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 2018, 35, 526–528. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Treffers, E.; Li, Y.; Tas, A.; Sun, Z.; van der Meer, Y.; de Ru, A.H.; van Veelen, P.; Atkins, J.; Snijder, E.J.; et al. Efficient -2 frameshifting by mammalian ribosomes to synthesize an additional arterivirus protein. Proc. Natl. Acad. Sci. USA 2012, 109, E2920–E2928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gulyaeva, A.; Dunowska, M.; Hoogendoorn, E.; Giles, J.; Samborskiy, D.; Gorbalenya, A.E. Domain Organization and Evolution of the Highly Divergent 5’ Coding Region of Genomes of Arteriviruses, Including the Novel Possum Nidovirus. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Snijder, E.J.; Wassenaar, A.L.; Spaan, W.J.; Gorbalenya, A.E. The arterivirus Nsp2 protease. An unusual cysteine protease with primary structure similarities to both papain-like and chymotrypsin-like proteases. J. Biol. Chem. 1995, 270, 16671–16676. [Google Scholar] [CrossRef] [Green Version]
- Archambault, D.; Balasuriya, U.B.; Rowland, R.R.; Yang, H.; Yoo, D. Animal arterivirus infections. Biomed. Res. Int. 2014, 2014, 303841. [Google Scholar] [CrossRef]
- Li, Y.; Tas, A.; Sun, Z.; Snijder, E.J.; Fang, Y. Proteolytic processing of the porcine reproductive and respiratory syndrome virus replicase. Virus Res. 2015, 202, 48–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Shang, P.; Shyu, D.; Carrillo, C.; Naraghi-Arani, P.; Jaing, C.J.; Renukaradhya, G.J.; Firth, A.E.; Snijder, E.J.; Fang, Y. Nonstructural proteins nsp2TF and nsp2N of porcine reproductive and respiratory syndrome virus (PRRSV) play important roles in suppressing host innate immune responses. Virology 2018, 517, 164–176. [Google Scholar] [CrossRef] [PubMed]
- Wassenaar, A.L.; Spaan, W.J.; Gorbalenya, A.E.; Snijder, E.J. Alternative proteolytic processing of the arterivirus replicase ORF1a polyprotein: Evidence that NSP2 acts as a cofactor for the NSP4 serine protease. J. Virol. 1997, 71, 9313–9322. [Google Scholar] [CrossRef] [Green Version]
- Monadjem, A. Rodents of Sub-Saharan Africa: A Biogeographic and Taxonomic Synthesis. 1092p. 2015. Available online: https://www-degruyter-com.kuleuven.e-bronnen.be/document/doi/10.1515/9783110301915/html (accessed on 22 December 2020).
- Vanmechelen, B.; Bletsa, M.; Laenen, L.; Lopes, A.R.; Vergote, V.; Beller, L.; Deboutte, W.; Korva, M.; Županc, T.A.; Gouy de Bellocq, J.; et al. Discovery and genome characterization of three new Jeilongviruses, a lineage of paramyxoviruses characterized by their unique membrane proteins. BMC Genom. 2018, 19, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Hooper, C.C.; Van Alstine, W.G.; Stevenson, G.W.; Kanitz, C.L. Mice and rats (laboratory and feral) are not a reservoir for PRRS virus. J. Vet. Diagn. Inv. 1994, 6, 13–15. [Google Scholar] [CrossRef]
- Cui, J.; Tachedjian, G.; Wang, L.-F. Bats and Rodents Shape Mammalian Retroviral Phylogeny. Sci. Rep. 2015, 5, 16561. [Google Scholar] [CrossRef]
- Souza, W.M.; Bello, G.; Amarilla, A.A.; Alfonso, H.L.; Aquino, V.H.; Figueiredo, L.T. Phylogeography and evolutionary history of rodent-borne hantaviruses. Infect. Genet. Evol. 2014, 21, 198–204. [Google Scholar] [CrossRef]
- Kuhn, J.H.; Lauck, M.; Bailey, A.L.; Shchetinin, A.M.; Vishnevskaya, T.V.; Bào, Y.; Ng, T.F.F.; LeBreton, M.; Schneider, B.S.; Gillis, A.; et al. Reorganization and expansion of the nidoviral family Arteriviridae. Arch. Virol. 2015, 161, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Arteriviridae Study Group. Expansion of the Rank Structure of the Family Arteriviridae and Renaming Its Taxa. 2017. Available online: https://talk.ictvonline.org/taxonomy/p/taxonomy-history?taxnode_id=20171822 (accessed on 2 December 2020).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Species | Positivity | Country of Origin |
|---|---|---|
| Acomys wilsoni | 0/1 | Tanzania |
| Graphiurus kelleni | 0/1 | Democratic Republic of the Congo |
| Lemniscomys striatus | 0/1 | Democratic Republic of the Congo |
| Lophuromys dudui | 0/7 | Democratic Republic of the Congo |
| Lophuromys laticeps | 0/3 | Tanzania |
| Lophuromys machangui | 3/16 | Mozambique, Tanzania |
| Lophuromys stanleyi | 0/4 | Tanzania |
| Mastomys natalensis | 1/1 | Tanzania |
| Micaelamys namaquensis | 0/1 | Mozambique |
| Praomys jacksoni | 2/5 | Democratic Republic of the Congo, Tanzania |
| Stenocephalemys albipes | 0/2 | Ethiopia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vanmechelen, B.; Zisi, Z.; Gryseels, S.; Goüy de Bellocq, J.; Vrancken, B.; Lemey, P.; Maes, P.; Bletsa, M. Phylogenomic Characterization of Lopma Virus and Praja Virus, Two Novel Rodent-Borne Arteriviruses. Viruses 2021, 13, 1842. https://doi.org/10.3390/v13091842
Vanmechelen B, Zisi Z, Gryseels S, Goüy de Bellocq J, Vrancken B, Lemey P, Maes P, Bletsa M. Phylogenomic Characterization of Lopma Virus and Praja Virus, Two Novel Rodent-Borne Arteriviruses. Viruses. 2021; 13(9):1842. https://doi.org/10.3390/v13091842
Chicago/Turabian StyleVanmechelen, Bert, Zafeiro Zisi, Sophie Gryseels, Joëlle Goüy de Bellocq, Bram Vrancken, Philippe Lemey, Piet Maes, and Magda Bletsa. 2021. "Phylogenomic Characterization of Lopma Virus and Praja Virus, Two Novel Rodent-Borne Arteriviruses" Viruses 13, no. 9: 1842. https://doi.org/10.3390/v13091842
APA StyleVanmechelen, B., Zisi, Z., Gryseels, S., Goüy de Bellocq, J., Vrancken, B., Lemey, P., Maes, P., & Bletsa, M. (2021). Phylogenomic Characterization of Lopma Virus and Praja Virus, Two Novel Rodent-Borne Arteriviruses. Viruses, 13(9), 1842. https://doi.org/10.3390/v13091842