
Role of Myeloid Cells in Oncolytic Reovirus-Based Cancer Therapy


Abstract
1. Introduction
2. Myeloid Cell Plasticity and the TME
2.1. M1 vs. M2 Macrophage Paradigm
2.2. Myeloid-Derived Suppressor Cells (MDSCs)
2.3. Tumor-Associated Macrophages (TAMs)
3. Reovirus-Based Dual-Prong Anticancer Actions and Myeloid Cells
3.1. Direct Oncolysis
3.2. Reovirus-Induced Antitumor Immunity
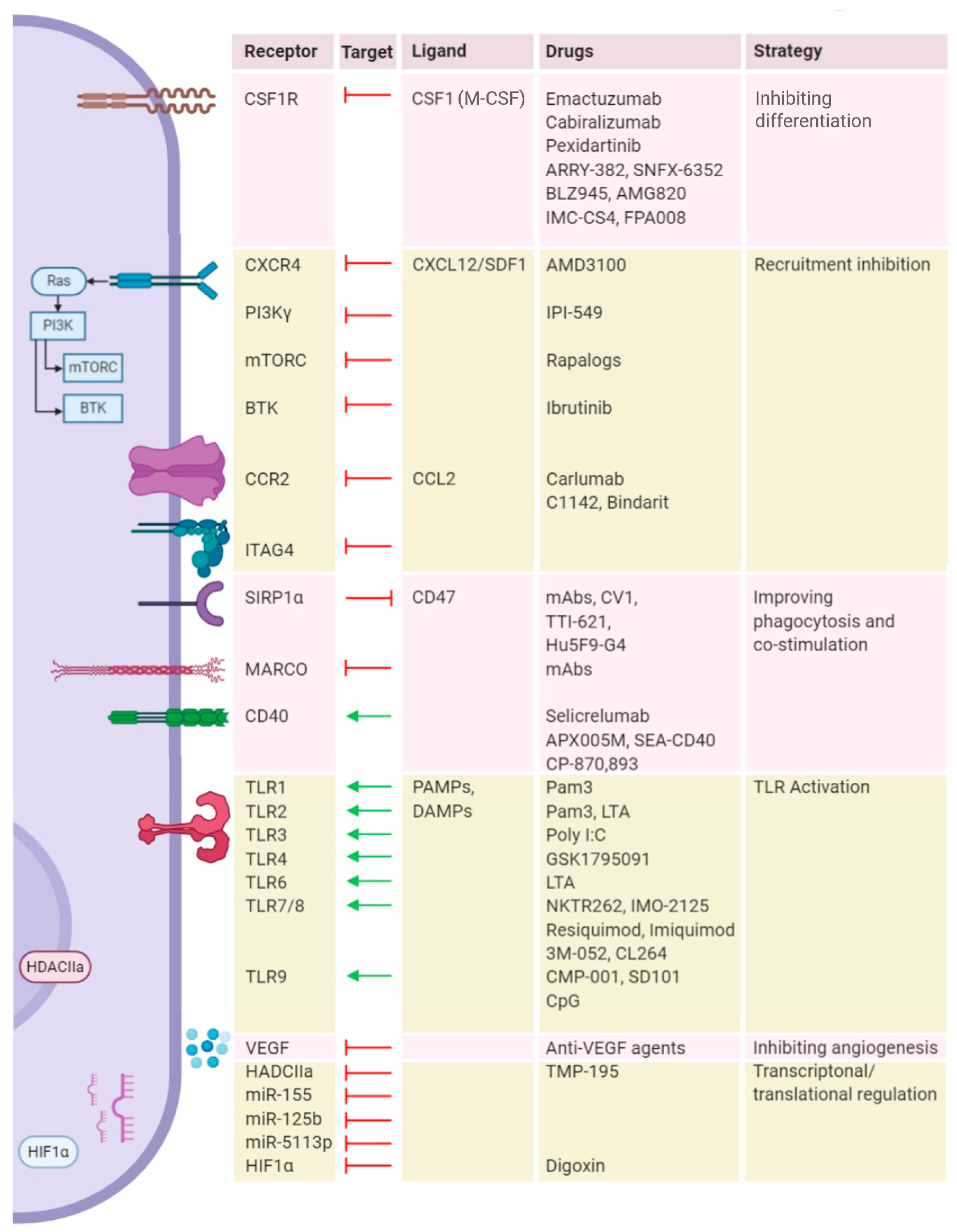
4. Targeting Myeloid Cells to Improve Reovirus Therapy
4.1. Blocking Recruitment
4.2. Depleting Macrophage Populations in the TME
4.3. Reprogramming Metabolism
4.4. Reprogramming Cellular Signaling
4.5. Immune Checkpoint Blockade (ICB)
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The tumor microenvironment and its role in promoting tumor growth. Oncogene 2008, 27, 5904–5912. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.; Pol, J.G.; Kroemer, G. Heating it up: Oncolytic viruses make tumors ‘hot’ and suitable for checkpoint blockade immunotherapies. Oncoimmunology 2018, 7, e1442169. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.P.; Kim, Y.; Clements, D.R.; Konda, P.; Schuster, H.; Kowalewski, D.J.; Paulo, J.A.; Cohen, A.M.; Stevanovic, S.; Gygi, S.P.; et al. Therapy-Induced MHC I Ligands Shape Neo-Antitumor CD8 T Cell Responses during Oncolytic Virus-Based Cancer Immunotherapy. J. Proteome Res. 2019, 18, 2666–2675. [Google Scholar] [CrossRef]
- Gujar, S.; Bell, J.; Diallo, J.S. SnapShot: Cancer Immunotherapy with Oncolytic Viruses. Cell 2019, 176, 1240. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.; Pol, J.G.; Kim, Y.; Lee, P.W.; Kroemer, G. Antitumor Benefits of Antiviral Immunity: An Underappreciated Aspect of Oncolytic Virotherapies. Trends Immunol. 2018, 39, 209–221. [Google Scholar] [CrossRef]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed]
- Maroun, J.; Muñoz-Alía, M.; Ammayappan, A.; Schulze, A.; Peng, K.W.; Russell, S. Designing and building oncolytic viruses. Future Virol. 2017, 12, 193–213. [Google Scholar] [CrossRef]
- Zhang, B.; Wang, X.; Cheng, P. Remodeling of Tumor Immune Microenvironment by Oncolytic Viruses. Front. Oncol. 2021, 10, 3478. [Google Scholar] [CrossRef]
- Kozak, R.; Hattin, L.; Biondi, M.; Corredor, J.; Walsh, S.; Xue-Zhong, M.; Manuel, J.; McGilvray, I.; Morgenstern, J.; Lusty, E.; et al. Replication and Oncolytic Activity of an Avian Orthoreovirus in Human Hepatocellular Carcinoma Cells. Viruses 2017, 9, 90. [Google Scholar] [CrossRef]
- Cai, R.; Meng, G.; Li, Y.; Wang, W.; Diao, Y.; Zhao, S.; Feng, Q.; Tang, Y. The oncolytic efficacy and safety of avian reovirus and its dynamic distribution in infected mice. Exp. Biol. Med. 2019, 244, 983–991. [Google Scholar] [CrossRef] [PubMed]
- Shmulevitz, M.; Marcato, P.; Lee, P.W.K. Unshackling the links between reovirus oncolysis, Ras signaling, translational control and cancer. Oncogene 2005, 24, 7720–7728. [Google Scholar] [CrossRef]
- Gujar, S.; Dielschneider, R.; Clements, D.; Helson, E.; Shmulevitz, M.; Marcato, P.; Pan, D.; Pan, L.Z.; Ahn, D.G.; Alawadhi, A.; et al. Multifaceted therapeutic targeting of ovarian peritoneal carcinomatosis through virus-induced immunomodulation. Mol. Ther. 2013, 21, 338–347. [Google Scholar] [CrossRef] [PubMed]
- Gujar, S.A.; Lee, P.W.K. Oncolytic virus-mediated reversal of impaired tumor antigen presentation. Front. Oncol. 2014, 4 APR, 77. [Google Scholar] [CrossRef]
- Gujar, S.A.; Clements, D.; Dielschneider, R.; Helson, E.; Marcato, P.; Lee, P.W.K. Gemcitabine enhances the efficacy of reovirus-based oncotherapy through anti-tumour immunological mechanisms. Br. J. Cancer 2014, 110, 83–93. [Google Scholar] [CrossRef]
- Gujar, S.A.; Marcato, P.; Pan, D.; Lee, P.W.K. Reovirus virotherapy overrides tumor antigen presentation evasion and promotes protective antitumor immunity. Mol. Cancer Ther. 2010, 9, 2924–2933. [Google Scholar] [CrossRef]
- White, C.L.; Twigger, K.R.; Vidal, L.; De Bono, J.S.; Coffey, M.; Heinemann, L.; Morgan, R.; Merrick, A.; Errington, F.; Vile, R.G.; et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther. 2008, 15, 911–920. [Google Scholar] [CrossRef]
- Ginhoux, F.; Jung, S. Monocytes and macrophages: Developmental pathways and tissue homeostasis. Nat. Rev. Immunol. 2014, 14, 392–404. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, D.; Iida, T.; Nakase, H. The Phagocytic Function of Macrophage-Enforcing Innate Immunity and Tissue Homeostasis. Int. J. Mol. Sci. 2017, 19, 92. [Google Scholar] [CrossRef]
- Zhou, J.; Tang, Z.; Gao, S.; Li, C.; Feng, Y.; Zhou, X. Tumor-Associated Macrophages: Recent Insights and Therapies. Front. Oncol. 2020, 10, 188. [Google Scholar] [CrossRef]
- Lacey, D.C.; Achuthan, A.; Fleetwood, A.J.; Dinh, H.; Roiniotis, J.; Scholz, G.M.; Chang, M.W.; Beckman, S.K.; Cook, A.D.; Hamilton, J.A. Defining GM-CSF– and Macrophage-CSF–Dependent Macrophage Responses by In Vitro Models. J. Immunol. 2012, 188, 5752–5765. [Google Scholar] [CrossRef]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000Prime Rep. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Ushach, I.; Zlotnik, A. Biological role of granulocyte macrophage colony-stimulating factor (GM-CSF) and macrophage colony-stimulating factor (M-CSF) on cells of the myeloid lineage. J. Leukoc. Biol. 2016, 100, 481–489. [Google Scholar] [CrossRef]
- Hamilton, T.A.; Zhao, C.; Pavicic, P.G.; Datta, S. Myeloid colony-stimulating factors as regulators of macrophage polarization. Front. Immunol. 2014, 5, 554. [Google Scholar] [CrossRef]
- Viola, A.; Munari, F.; Sánchez-Rodríguez, R.; Scolaro, T.; Castegna, A. The metabolic signature of macrophage responses. Front. Immunol. 2019, 10, 1462. [Google Scholar] [CrossRef]
- O’Neill, L.A.J. A Metabolic Roadblock in Inflammatory Macrophages. Cell Rep. 2016, 17, 625–626. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Fares, C.M.; Van Allen, E.M.; Drake, C.G.; Allison, J.P.; Hu-Lieskovan, S. Mechanisms of Resistance to Immune Checkpoint Blockade: Why Does Checkpoint Inhibitor Immunotherapy Not Work for All Patients? Am. Soc. Clin. Oncol. Educ. Book. Am. Soc. Clin. Oncol. Annu. Meet. 2019, 39, 147–164. [Google Scholar] [CrossRef] [PubMed]
- Pender, A.; Titmuss, E.; Pleasance, E.D.; Fan, K.Y.; Pearson, H.; Brown, S.D.; Grisdale, C.J.; Topham, J.T.; Shen, Y.; Bonakdar, M.; et al. Genome and transcriptome biomarkers of response to immune checkpoint inhibitors in advanced solid tumors. Clin. Cancer Res. 2021, 27, 202–212. [Google Scholar] [CrossRef] [PubMed]
- Jayasingam, S.D.; Citartan, M.; Thang, T.H.; Mat Zin, A.A.; Ang, K.C.; Ch’ng, E.S. Evaluating the Polarization of Tumor-Associated Macrophages Into M1 and M2 Phenotypes in Human Cancer Tissue: Technicalities and Challenges in Routine Clinical Practice. Front. Oncol. 2020, 9, 1512. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; DeNardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-Based Network Analysis Reveals a Spectrum Model of Human Macrophage Activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef]
- Kiss, M.; Van Gassen, S.; Movahedi, K.; Saeys, Y.; Laoui, D. Myeloid cell heterogeneity in cancer: Not a single cell alike. Cell. Immunol. 2018, 330, 188–201. [Google Scholar] [CrossRef]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity, Mechanisms, and Significance of Macrophage Plasticity. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 123–147. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [PubMed]
- Schmid, M.C.; Franco, I.; Kang, S.W.; Hirsch, E.; Quilliam, L.A.; Varner, J.A. PI3-Kinase γ Promotes Rap1a-Mediated Activation of Myeloid Cell Integrin α4β1, Leading to Tumor Inflammation and Growth. PLoS ONE 2013, 8, e60226. [Google Scholar] [CrossRef] [PubMed]
- Lesokhin, A.M.; Hohl, T.M.; Kitano, S.; Cortez, C.; Hirschhorn-Cymerman, D.; Avogadri, F.; Rizzuto, G.A.; Lazarus, J.J.; Pamer, E.G.; Houghton, A.N.; et al. Monocytic CCR2 + myeloid-derived suppressor cells promote immune escape by limiting activated CD8 T-cell infiltration into the tumor microenvironment. Cancer Res. 2012, 72, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Condamine, T.; Ramachandran, I.; Youn, J.-I.; Gabrilovich, D.I. Regulation of Tumor Metastasis by Myeloid-Derived Suppressor Cells. Annu. Rev. Med. 2015, 66, 97–110. [Google Scholar] [CrossRef] [PubMed]
- Gabrilovich, D.I. Myeloid-derived suppressor cells. Cancer Immunol. Res. 2017, 5, 3–8. [Google Scholar] [CrossRef]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef]
- Condamine, T.; Gabrilovich, D.I. Molecular mechanisms regulating myeloid-derived suppressor cell differentiation and function. Trends Immunol. 2011, 32, 19–25. [Google Scholar] [CrossRef]
- Davidov, V.; Jensen, G.; Mai, S.; Chen, S.H.; Pan, P.Y. Analyzing One Cell at a TIME: Analysis of Myeloid Cell Contributions in the Tumor Immune Microenvironment. Front. Immunol. 2020, 11, 1842. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.F.; Song, S.Y.; Wang, T.J.; Ji, W.J.; Li, S.W.; Liu, N.; Yan, C.X. Prognostic role of pretreatment circulating MDSCs in patients with solid malignancies: A meta-analysis of 40 studies. Oncoimmunology 2018, 7, e1494113. [Google Scholar] [CrossRef] [PubMed]
- Fultang, N.; Li, X.; Li, T.; Chen, Y.H. Myeloid-Derived Suppressor Cell Differentiation in Cancer: Transcriptional Regulators and Enhanceosome-Mediated Mechanisms. Front. Immunol. 2021, 11, 3493. [Google Scholar] [CrossRef]
- Beury, D.W.; Parker, K.H.; Nyandjo, M.; Sinha, P.; Carter, K.A.; Ostrand-Rosenberg, S. Cross-talk among myeloid-derived suppressor cells, macrophages, and tumor cells impacts the inflammatory milieu of solid tumors. J. Leukoc. Biol. 2014, 96, 1109–1118. [Google Scholar] [CrossRef]
- Murdoch, C.; Muthana, M.; Coffelt, S.B.; Lewis, C.E. The role of myeloid cells in the promotion of tumour angiogenesis. Nat. Rev. Cancer 2008, 8, 618–631. [Google Scholar] [CrossRef]
- Petty, A.J.; Yang, Y. Tumor-associated macrophages: Implications in cancer immunotherapy. Immunotherapy 2017, 9, 289–302. [Google Scholar] [CrossRef]
- Guerriero, J.L. Macrophages: The Road Less Traveled, Changing Anticancer Therapy. Trends Mol. Med. 2018, 24, 472–489. [Google Scholar] [CrossRef] [PubMed]
- Noy, R.; Pollard, J.W. Tumor-Associated Macrophages: From Mechanisms to Therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and Metabolism in the Tumor Microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef] [PubMed]
- Kelly, P.M.A.; Davison, R.S.; Bliss, E.; McGee, J.O.D. Macrophages in human breast disease: A quantitative immunohistochemical study. Br. J. Cancer 1988, 57, 174–177. [Google Scholar] [CrossRef]
- Hanada, T.; Nakagawa, M.; Emoto, A.; Nomura, T.; Nasu, N.; Nomura, Y. Prognostic value of tumor-associated macrophage count in human bladder cancer. Int. J. Urol. 2000, 7, 263–269. [Google Scholar] [CrossRef]
- Ciavarra, R.P.; Taylor, L.; Greene, A.R.; Yousefieh, N.; Horeth, D.; Van Rooijen, N.; Steel, C.; Gregory, B.; Birkenbach, M.; Sekellick, M. Impact of macrophage and dendritic cell subset elimination on antiviral immunity, viral clearance and production of type 1 interferon. Virology 2005, 342, 177–189. [Google Scholar] [CrossRef]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Bronte, V. Altered macrophage differentiation and immune dysfunction in tumor development. J. Clin. Investig. 2007, 117, 1155–1166. [Google Scholar] [CrossRef] [PubMed]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef] [PubMed]
- Saeys, Y.; Van Gassen, S.; Lambrecht, B.N. Computational flow cytometry: Helping to make sense of high-dimensional immunology data. Nat. Rev. Immunol. 2016, 16, 449–462. [Google Scholar] [CrossRef]
- Cassetta, L.; Fragkogianni, S.; Sims, A.H.; Swierczak, A.; Forrester, L.M.; Zhang, H.; Soong, D.Y.H.; Cotechini, T.; Anur, P.; Lin, E.Y.; et al. Human Tumor-Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019, 35, 588–602. [Google Scholar] [CrossRef]
- Zhu, X.D.; Zhang, J.B.; Zhuang, P.Y.; Zhu, H.G.; Zhang, W.; Xiong, Y.Q.; Wu, W.Z.; Wang, L.; Tang, Z.Y.; Sun, H.C. High expression of macrophage colony-stimulating factor in peritumoral liver tissue is associated with poor survival after curative resection of hepatocellular carcinoma. J. Clin. Oncol. 2008, 26, 2707–2716. [Google Scholar] [CrossRef]
- Lin, E.Y.; Nguyen, A.V.; Russell, R.G.; Pollard, J.W. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J. Exp. Med. 2001, 193, 727–739. [Google Scholar] [CrossRef]
- Ruffell, B.; Affara, N.I.; Coussens, L.M. Differential macrophage programming in the tumor microenvironment. Trends Immunol. 2012, 33, 119–126. [Google Scholar] [CrossRef]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef]
- Qian, B.Z.; Pollard, J.W. Macrophage Diversity Enhances Tumor Progression and Metastasis. Cell 2010, 141, 39–51. [Google Scholar] [CrossRef]
- Mizutani, K.; Sud, S.; McGregor, N.A.; Martinovski, G.; Rice, B.T.; Craig, M.J.; Varsos, Z.S.; Roca, H.; Pienta, K.J. The chemokine CCL2 increases prostate tumor growth and bone metastasis through macrophage and osteoclast recruitment. Neoplasia 2009, 11, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Serafini, P.; Carbley, R.; Noonan, K.A.; Tan, G.; Bronte, V.; Borrello, I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004, 64, 6337–6343. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Sun, J.; Kim, S.; Kapoor, V.; Cheng, G.; Ling, L.; Worthen, G.S.; Albelda, S.M. Polarization of Tumor-Associated Neutrophil Phenotype by TGF-β: “N1” versus “N2” TAN. Cancer Cell 2009, 16, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Jiffry, J.; Thavornwatanayong, T.; Rao, D.; Fogel, E.J.; Saytoo, D.; Nahata, R.; Guzik, H.; Chaudhary, I.; Augustine, T.; Goel, S.; et al. Oncolytic reovirus (pelareorep) induces autophagy in KRAS-mutated colorectal cancer. Clin. Cancer Res. 2021, 27, 865–876. [Google Scholar] [CrossRef]
- Maitra, R.; Seetharam, R.; Tesfa, L.; Augustine, T.A.; Klampfer, L.; Coffey, M.C.; Mariadason, J.M.; Goel, S. Oncolytic reovirus preferentially induces apoptosis in KRAS mutant colorectal cancer cells, and synergizes with irinotecan. Oncotarget 2014, 5, 2807–2819. [Google Scholar] [CrossRef]
- Villalona-Calero, M.A.; Lam, E.; Otterson, G.A.; Zhao, W.; Timmons, M.; Subramaniam, D.; Hade, E.M.; Gill, G.M.; Coffey, M.; Selvaggi, G.; et al. Oncolytic reovirus in combination with chemotherapy in metastatic or recurrent non-small cell lung cancer patients with KRAS-activated tumors. Cancer 2016, 122, 875–883. [Google Scholar] [CrossRef]
- Danthi, P.; Guglielmi, K.M.; Kirchner, E.; Mainou, B.; Stehle, T.; Dermody, T.S. From Touchdown to Transcription: The Reovirus Cell Entry Pathway. In Cell Entry by Non-Enveloped Viruses; Springer: Berlin, Heidelberg, 2010; Volume 343, pp. 91–119. [Google Scholar]
- Müller, L.; Berkeley, R.; Barr, T.; Ilett, E.; Errington-Mais, F. Past, Present and Future of Oncolytic Reovirus. Cancers (Basel) 2020, 12, 3219. [Google Scholar] [CrossRef]
- Galluzzi, L.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Agostinis, P.; Alnemri, E.S.; Altucci, L.; Amelio, I.; Andrews, D.W.; et al. Molecular mechanisms of cell death: Recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018, 25, 486–541. [Google Scholar] [CrossRef]
- Ikeda, Y.; Nishimura, G.; Yanoma, S.; Kubota, A.; Furukawa, M.; Tsukuda, M. Reovirus oncolysis in human head and neck squamous carcinoma cells. Auris Nasus Larynx 2004, 31, 407–412. [Google Scholar] [CrossRef]
- Yuan, J.; Kroemer, G. Alternative cell death mechanisms in development and beyond. Genes Dev. 2010, 24, 2592–2602. [Google Scholar] [CrossRef]
- Thirukkumaran, C.; Morris, D.G. Oncolytic viral therapy using reovirus. Methods Mol. Biol. 2015, 1317, 187–223. [Google Scholar] [CrossRef] [PubMed]
- Giacomantonio, M.A.; Sterea, A.M.; Kim, Y.; Paulo, J.A.; Clements, D.R.; Kennedy, B.E.; Bydoun, M.J.; Shi, G.; Waisman, D.M.; Gygi, S.P.; et al. Quantitative Proteome Responses to Oncolytic Reovirus in GM-CSF-and M-CSF-Differentiated Bone Marrow-Derived Cells. J. Proteome Res. 2020, 19, 708–718. [Google Scholar] [CrossRef] [PubMed]
- Clements, D.R.; Murphy, J.P.; Sterea, A.; Kennedy, B.E.; Kim, Y.; Helson, E.; Almasi, S.; Holay, N.; Konda, P.; Paulo, J.A.; et al. Quantitative Temporal in Vivo Proteomics Deciphers the Transition of Virus-Driven Myeloid Cells into M2 Macrophages. J. Proteome Res. 2017, 16, 3391–3406. [Google Scholar] [CrossRef] [PubMed]
- Fulci, G.; Breymann, L.; Gianni, D.; Kurozomi, K.; Rhee, S.S.; Yu, J.; Kaur, B.; Louis, D.N.; Weissleder, R.; Caligiuri, M.A.; et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12873–12878. [Google Scholar] [CrossRef] [PubMed]
- Tan, Z.; Liu, L.; Chiu, M.S.; Cheung, K.W.; Yan, C.W.; Yu, Z.; Lee, B.K.; Liu, W.; Man, K.; Chen, Z. Virotherapy-recruited PMN-MDSC infiltration of mesothelioma blocks antitumor CTL by IL-10-mediated dendritic cell suppression. Oncoimmunology 2019, 8, e1518672. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, B.E.; Sadek, M.; Gujar, S.A. Targeted Metabolic Reprogramming to Improve the Efficacy of Oncolytic Virus Therapy. Mol. Ther. 2020, 28, 1417–1421. [Google Scholar] [CrossRef] [PubMed]
- Farhood, B.; Najafi, M.; Mortezaee, K. CD8+ cytotoxic T lymphocytes in cancer immunotherapy: A review. J. Cell. Physiol. 2019, 234, 8509–8521. [Google Scholar] [CrossRef]
- Clements, D.R.; Sterea, A.M.; Kim, Y.; Helson, E.; Dean, C.A.; Nunokawa, A.; Coyle, K.M.; Sharif, T.; Marcato, P.; Gujar, S.A.; et al. Newly Recruited CD11b +, GR-1 +, Ly6C high Myeloid Cells Augment Tumor-Associated Immunosuppression Immediately following the Therapeutic Administration of Oncolytic Reovirus. J. Immunol. 2015, 194, 4397–4412. [Google Scholar] [CrossRef]
- Gujar, S.A.; Clements, D.; Lee, P.W.K. Two is better than one: Complementing oncolytic virotherapy with gemcitabine to potentiate antitumor immune responses. Oncoimmunology 2014, 3. [Google Scholar] [CrossRef]
- Katayama, Y.; Tachibana, M.; Kurisu, N.; Oya, Y.; Terasawa, Y.; Goda, H.; Kobiyama, K.; Ishii, K.J.; Akira, S.; Mizuguchi, H.; et al. Oncolytic Reovirus Inhibits Immunosuppressive Activity of Myeloid-Derived Suppressor Cells in a TLR3-Dependent Manner. J. Immunol. 2018, 200, 2987–2999. [Google Scholar] [CrossRef]
- Parker, J.N.; Gillespie, G.Y.; Love, C.E.; Randall, S.; Whitley, R.J.; Markert, J.M. Engineered herpes simplex virus expressing IL-12 in the treatment of experimental murine brain tumors. Proc. Natl. Acad. Sci. USA 2000, 97, 2208–2213. [Google Scholar] [CrossRef]
- Kleijn, A.; Kloezeman, J.; Treffers-Westerlaken, E.; Fulci, G.; Leenstra, S.; Dirven, C.; Debets, R.; Lamfers, M. The In Vivo Therapeutic Efficacy of the Oncolytic Adenovirus Delta24-RGD Is Mediated by Tumor-Specific Immunity. PLoS ONE 2014, 9, e97495. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.Q.; Zhang, L.; Ohba, K.; Ye, M.; Ichiyama, K.; Yamamoto, N. Macrophage response to oncolytic paramyxoviruses potentiates virus-mediated tumor cell killing. Eur. J. Immunol. 2016, 46, 919–928. [Google Scholar] [CrossRef]
- Esaki, S.; Goshima, F.; Kimura, H.; Murakami, S.; Nishiyama, Y. Enhanced antitumoral activity of oncolytic herpes simplex virus with gemcitabine using colorectal tumor models. Int. J. Cancer 2013, 132, 1592–1601. [Google Scholar] [CrossRef] [PubMed]
- Togashi, Y.; Shitara, K.; Nishikawa, H. Regulatory T cells in cancer immunosuppression—Implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019, 16, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Siret, C.; Collignon, A.; Silvy, F.; Robert, S.; Cheyrol, T.; André, P.; Rigot, V.; Iovanna, J.; van de Pavert, S.; Lombardo, D.; et al. Deciphering the Crosstalk Between Myeloid-Derived Suppressor Cells and Regulatory T Cells in Pancreatic Ductal Adenocarcinoma. Front. Immunol. 2020, 10, 3070. [Google Scholar] [CrossRef] [PubMed]
- Lucca, L.E.; Dominguez-Villar, M. Modulation of regulatory T cell function and stability by co-inhibitory receptors. Nat. Rev. Immunol. 2020, 20, 680–693. [Google Scholar] [CrossRef]
- Savage, P.A.; Klawon, D.E.J.; Miller, C.H. Regulatory T Cell Development. Annu. Rev. Immunol. 2020, 38, 421–453. [Google Scholar] [CrossRef] [PubMed]
- Qian, B.Z.; Li, J.; Zhang, H.; Kitamura, T.; Zhang, J.; Campion, L.R.; Kaiser, E.A.; Snyder, L.A.; Pollard, J.W. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature 2011, 475, 222–225. [Google Scholar] [CrossRef] [PubMed]
- Ren, G.; Zhao, X.; Wang, Y.; Zhang, X.; Chen, X.; Xu, C.; Yuan, Z.R.; Roberts, A.I.; Zhang, L.; Zheng, B.; et al. CCR2-dependent recruitment of macrophages by tumor-educated mesenchymal stromal cells promotes tumor development and is mimicked by TNFα. Cell Stem Cell 2012, 11, 812–824. [Google Scholar] [CrossRef] [PubMed]
- Fridlender, Z.G.; Buchlis, G.; Kapoor, V.; Cheng, G.; Sun, J.; Singhal, S.; Crisanti, C.; Wang, L.C.S.; Heitjan, D.; Snyder, L.A.; et al. CCL2 blockade augments cancer immunotherapy. Cancer Res. 2010, 70, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Bonapace, L.; Coissieux, M.M.; Wyckoff, J.; Mertz, K.D.; Varga, Z.; Junt, T.; Bentires-Alj, M. Cessation of CCL2 inhibition accelerates breast cancer metastasis by promoting angiogenesis. Nature 2014, 515, 130–133. [Google Scholar] [CrossRef] [PubMed]
- Pathria, P.; Louis, T.L.; Varner, J.A. Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 2019, 40, 310–327. [Google Scholar] [CrossRef] [PubMed]
- Hughes, R.; Qian, B.Z.; Rowan, C.; Muthana, M.; Keklikoglou, I.; Olson, O.C.; Tazzyman, S.; Danson, S.; Addison, C.; Clemons, M.; et al. Perivascular M2 macrophages stimulate tumor relapse after chemotherapy. Cancer Res. 2015, 75, 3479–3491. [Google Scholar] [CrossRef]
- Scala, S. Molecular pathways: Targeting the CXCR4-CXCL12 Axis-Untapped potential in the tumor microenvironment. Clin. Cancer Res. 2015, 21, 4278–4285. [Google Scholar] [CrossRef]
- Teicher, B.A.; Fricker, S.P. CXCL12 (SDF-1)/CXCR4 pathway in cancer. Clin. Cancer Res. 2010, 16, 2927–2931. [Google Scholar] [CrossRef]
- Zheng, X.; Turkowski, K.; Mora, J.; Brüne, B.; Seeger, W.; Weigert, A.; Savai, R. Redirecting tumor-associated macrophages to become tumoricidal effectors as a novel strategy for cancer therapy. Oncotarget 2017, 8, 48436–48452. [Google Scholar] [CrossRef]
- Rogers, T.L.; Holen, I. Tumour macrophages as potential targets of bisphosphonates. J. Transl. Med. 2011, 9. [Google Scholar] [CrossRef] [PubMed]
- Giraudo, E.; Inoue, M.; Hanahan, D. An amino-bisphosphonate targets MMP-9–expressing macrophages and angiogenesis to impair cervical carcinogenesis. J. Clin. Investig. 2004, 114, 623–633. [Google Scholar] [CrossRef]
- Germano, G.; Frapolli, R.; Belgiovine, C.; Anselmo, A.; Pesce, S.; Liguori, M.; Erba, E.; Uboldi, S.; Zucchetti, M.; Pasqualini, F.; et al. Role of Macrophage Targeting in the Antitumor Activity of Trabectedin. Cancer Cell 2013, 23, 249–262. [Google Scholar] [CrossRef]
- Denton, N.L.; Chen, C.Y.; Hutzen, B.; Currier, M.A.; Scott, T.; Nartker, B.; Leddon, J.L.; Wang, P.Y.; Srinivas, R.; Cassady, K.A.; et al. Myelolytic Treatments Enhance Oncolytic Herpes Virotherapy in Models of Ewing Sarcoma by Modulating the Immune Microenvironment. Mol. Ther. Oncolytics 2018, 11, 62–74. [Google Scholar] [CrossRef]
- Fulci, G.; Dmitrieva, N.; Gianni, D.; Fontana, E.J.; Pan, X.; Lu, Y.; Kaufman, C.S.; Kaur, B.; Lawler, S.E.; Lee, R.J.; et al. Depletion of peripheral macrophages and brain microglia increases brain tumor titers of oncolytic viruses. Cancer Res. 2007, 67, 9398–9406. [Google Scholar] [CrossRef]
- O’Neill, L.A.J. A Broken Krebs Cycle in Macrophages. Immunity 2015, 42, 393–394. [Google Scholar] [CrossRef]
- Van den Bossche, J.; O’Neill, L.A.; Menon, D. Macrophage Immunometabolism: Where Are We (Going)? Trends Immunol. 2017, 38, 395–406. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef]
- Jha, A.K.; Huang, S.C.C.; Sergushichev, A.; Lampropoulou, V.; Ivanova, Y.; Loginicheva, E.; Chmielewski, K.; Stewart, K.M.; Ashall, J.; Everts, B.; et al. Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 2015, 42, 419–430. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, J.; Baardman, J.; Otto, N.A.; van der Velden, S.; Neele, A.E.; van den Berg, S.M.; Luque-Martin, R.; Chen, H.J.; Boshuizen, M.C.S.; Ahmed, M.; et al. Mitochondrial Dysfunction Prevents Repolarization of Inflammatory Macrophages. Cell Rep. 2016, 17, 684–696. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.C.C.; Smith, A.M.; Everts, B.; Colonna, M.; Pearce, E.L.; Schilling, J.D.; Pearce, E.J. Metabolic Reprogramming Mediated by the mTORC2-IRF4 Signaling Axis Is Essential for Macrophage Alternative Activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef]
- Kowal, J.; Kornete, M.; Joyce, J.A. Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy 2019, 11, 677–689. [Google Scholar] [CrossRef]
- Barclay, A.N.; Van Den Berg, T.K. The interaction between signal regulatory protein alpha (SIRPα) and CD47: Structure, function, and therapeutic target. Annu. Rev. Immunol. 2014, 32, 25–50. [Google Scholar] [CrossRef]
- Weiskopf, K.; Ring, A.M.; Ho, C.C.M.; Volkmer, J.P.; Levin, A.M.; Volkmer, A.K.; Özkan, E.; Fernhoff, N.B.; Van De Rijn, M.; Weissman, I.L.; et al. Engineered SIRPα variants as immunotherapeutic adjuvants to anticancer antibodies. Science 2013, 341, 88–91. [Google Scholar] [CrossRef]
- Shi, G.; Yang, Q.; Zhang, Y.; Jiang, Q.; Lin, Y.; Yang, S.; Wang, H.; Cheng, L.; Zhang, X.; Li, Y.; et al. Modulating the Tumor Microenvironment via Oncolytic Viruses and CSF-1R Inhibition Synergistically Enhances Anti-PD-1 Immunotherapy. Mol. Ther. 2019, 27, 244–260. [Google Scholar] [CrossRef]
- Cerullo, V.; Diaconu, I.; Romano, V.; Hirvinen, M.; Ugolini, M.; Escutenaire, S.; Holm, S.L.; Kipar, A.; Kanerva, A.; Hemminki, A. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol. Ther. 2012, 20, 2076–2086. [Google Scholar] [CrossRef]
- Eriksson, E.; Moreno, R.; Milenova, I.; Liljenfeldt, L.; Dieterich, L.C.; Christiansson, L.; Karlsson, H.; Ullenhag, G.; Mangsbo, S.M.; Dimberg, A.; et al. Activation of myeloid and endothelial cells by CD40L gene therapy supports T-cell expansion and migration into the tumor microenvironment. Gene Ther. 2017, 24, 92–103. [Google Scholar] [CrossRef]
- Kemp, V.; van den Wollenberg, D.J.M.; Camps, M.G.M.; van Hall, T.; Kinderman, P.; Pronk-van Montfoort, N.; Hoeben, R.C. Arming oncolytic reovirus with GM-CSF gene to enhance immunity. Cancer Gene Ther. 2019, 26, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Breitbach, C.; Bell, J.C.; Hwang, T.-H.; Kirn, D.; Burke, J. The emerging therapeutic potential of the oncolytic immunotherapeutic Pexa-Vec (JX-594). Oncolytic Virotherapy 2015, 4, 25. [Google Scholar] [CrossRef]
- Park, S.H.; Breitbach, C.J.; Lee, J.; Park, J.O.; Lim, H.Y.; Kang, W.K.; Moon, A.; Mun, J.H.; Sommermann, E.M.; Maruri Avidal, L.; et al. Phase 1b Trial of Biweekly Intravenous Pexa-Vec (JX-594), an Oncolytic and Immunotherapeutic Vaccinia Virus in Colorectal Cancer. Mol. Ther. 2015, 23, 1532–1540. [Google Scholar] [CrossRef]
- Cripe, T.P.; Ngo, M.C.; Geller, J.I.; Louis, C.U.; Currier, M.A.; Racadio, J.M.; Towbin, A.J.; Rooney, C.M.; Pelusio, A.; Moon, A.; et al. Phase 1 study of intratumoral Pexa-Vec (JX-594), an oncolytic and immunotherapeutic vaccinia virus, in pediatric cancer patients. Mol. Ther. 2015, 23, 602–608. [Google Scholar] [CrossRef]
- Koch, M.; Lawler, S.; Chiocca, E. HSV-1 Oncolytic Viruses from Bench to Bedside: An Overview of Current Clinical Trials. Cancers (Basel) 2020, 12, 3514. [Google Scholar] [CrossRef]
- Middleton, M.R.; Aroldi, F.; Sacco, J.; Milhem, M.M.; Curti, B.D.; Vanderwalde, A.M.; Baum, S.; Samson, A.; Pavlick, A.C.; Chesney, J.A.; et al. An open-label, single-arm, phase II clinical trial of RP1, an enhanced potency oncolytic herpes virus, combined with nivolumab in four solid tumor types: Initial results from the skin cancer cohorts. J. Clin. Oncol. 2020, 38, e22050. [Google Scholar] [CrossRef]
- Pol, J.G.; Lévesque, S.; Workenhe, S.T.; Gujar, S.; Le Boeuf, F.; Clements, D.R.; Fahrner, J.E.; Fend, L.; Bell, J.C.; Mossman, K.L.; et al. Trial Watch: Oncolytic viro-immunotherapy of hematologic and solid tumors. Oncoimmunology 2018, 7. [Google Scholar] [CrossRef]
- Pol, J.; Kroemer, G.; Galluzzi, L. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology 2016, 5, e1115641. [Google Scholar] [CrossRef]
- Liu, B.L.; Robinson, M.; Han, Z.Q.; Branston, R.H.; English, C.; Reay, P.; McGrath, Y.; Thomas, S.K.; Thornton, M.; Bullock, P.; et al. ICP34.5 deleted herpes simplex virus with enhanced oncolytic, immune stimulating, and anti-tumour properties. Gene Ther. 2003, 10, 292–303. [Google Scholar] [CrossRef]
- Liu, M.; Zhou, J.; Liu, X.; Feng, Y.; Yang, W.; Wu, F.; Cheung, O.K.W.; Sun, H.; Zeng, X.; Tang, W.; et al. Targeting monocyte-intrinsic enhancer reprogramming improves immunotherapy efficacy in hepatocellular carcinoma. Gut 2020, 69, 365–379. [Google Scholar] [CrossRef]
- Chen, J.; Sun, H.W.; Yang, Y.Y.; Chen, H.T.; Yu, X.J.; Wu, W.C.; Xu, Y.T.; Jin, L.L.; Wu, X.J.; Xu, J.; et al. Reprogramming immunosuppressive myeloid cells by activated T cells promotes the response to anti-PD-1 therapy in colorectal cancer. Signal Transduct. Target. Ther. 2021, 6, 1–14. [Google Scholar] [CrossRef]
- Pico De Coaña, Y.; Masucci, G.; Hansson, J.; Kiessling, R. Myeloid-derived suppressor cells and their role in CTLA-4 blockade therapy. Cancer Immunol. Immunother. 2014, 63, 977–983. [Google Scholar] [CrossRef]
- Chesney, J.A.; Mitchell, R.A.; Yaddanapudi, K. Myeloid-derived suppressor cells—a new therapeutic target to overcome resistance to cancer immunotherapy. J. Leukoc. Biol. 2017, 102, 727–740. [Google Scholar] [CrossRef]
- Bar, N.; Costa, F.; Das, R.; Duffy, A.; Samur, M.; McCachren, S.; Gettinger, S.N.; Neparidze, N.; Parker, T.L.; Bailur, J.K.; et al. Differential effects of PD-L1 versus PD-1 blockade on myeloid inflammation in human cancer. JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Cannarile, M.A.; Weisser, M.; Jacob, W.; Jegg, A.M.; Ries, C.H.; Rüttinger, D. Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 2017, 5, 53. [Google Scholar] [CrossRef]
- Wiehagen, K.R.; Girgis, N.M.; Yamada, D.H.; Smith, A.A.; Chan, S.R.; Grewal, I.S.; Quigley, M.; Verona, R.I. Combination of CD40 agonism and CSF-1R blockade reconditions tumor-associated macrophages and drives potent antitumor immunity. Cancer Immunol. Res. 2017, 5, 1109–1121. [Google Scholar] [CrossRef]
- Weber, A.N.R.; Bittner, Z.; Liu, X.; Dang, T.M.; Radsak, M.P.; Brunner, C. Bruton’s tyrosine kinase: An emerging key player in innate immunity. Front. Immunol. 2017, 8, 1454. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, Q.; Xu, M.; Wang, L.; Chen, X.; Feng, Y.; Li, Y.; Zhang, X.; Cui, W.; Jia, X. CCL2-CCR2 axis recruits tumor associated macrophages to induce immune evasion through PD-1 signaling in esophageal carcinogenesis. Mol. Cancer 2020, 19, 41. [Google Scholar] [CrossRef]
- Wang, W.; Marinis, J.M.; Beal, A.M.; Savadkar, S.; Wu, Y.; Khan, M.; Taunk, P.S.; Wu, N.; Su, W.; Wu, J.; et al. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 2018, 34, 757–774. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Trial | Virus Type | OV Agent | Immunomodulatory Therapy | Phase (s) | Cancer (s) | |
|---|---|---|---|---|---|---|
| Agent | Description | |||||
| NCT03747744 | HSV | T-Vec | CD1c (BDCA-1)+ myDC | CD1c+ myeloid dendritic cells | I | Melanoma |
| NCT02197169 | Adenovirus | DNX-2401 | IFNγ | Pro-inflammatory | I | Glioblastoma or Gliosarcoma |
| NCT02143804 | Adenovirus | CG0070 | GM-CSF (encoded) | M1 polarizer | II | Bladder Cancer, High Grade, Non-Muscle Invasive |
| NCT00625456 | Vaccinia | RACVAC (JX-594) | GM-CSF (encoded) | M1 polarizer | I | Melanoma, Lung Cancer Renal Cell Carcinoma, Squamous Cell Carcinoma of the Head and Neck, Neuroblastoma |
| NCT01169584 | Vaccinia | RACVAC (JX-594) | GM-CSF (encoded) | M1 polarizer | I | Rhabdomyosarcoma, Lymphoma Wilm’s Tumor, Ewing’s Sarcoma |
| NCT04725331 | Vaccinia | BT-001 | Anti CTLA-4 mAb (encoded) | ICB | I, II | Solid Tumor, Adult Metastatic Cancer Soft Tissue Sarcoma, Merkel Cell Carcinoma, Melanoma, Triple Negative Breast Cancer Non-Small Cell Lung Cancer |
| NCT04050436 | HSV | RP1 | GM-CSF (encoded) | M1 polarizer | II | Squamous Cell Carcinoma |
| Cemiplimab (combination) | PD-1 | |||||
| NCT03767348 | HSV | RP1 | GM-CSF (encoded) | M1 polarizer | II | Melanoma, NSCLC |
| Nivolumab (combination) | PD-1 | II | ||||
| NCT00554372 | Vaccinia | JX-594 | GM-CSF (encoded) | M1 polarizer | II | Hepatocellular Carcinoma |
| NCT00629759 | Vaccinia | JX-594 | GM-CSF (encoded) | M1 polarizer | I | Liver neoplasms |
| NCT04521764 | Measles | MV | NAP (encoded) | Secretes neutrophil activating protein | I | Breast cancer |
| Trial | Reoviral Agent | ICB therapy | Phase (s) | Cancer (s) | |
|---|---|---|---|---|---|
| Agent | Target | ||||
| NCT03723915 | Pelareorep | Pembrolizumab | PD-1 | II | Pancreatic adenocarcinoma, Pancreatic cancer |
| NCT03605719 | Pelareorep | Nivolumab | PD-1 | I | Recurrent Plasma Cell Myeloma |
| NCT04102618 | Pelareorep | Atezolizumab | PD-L1 | I | Breast cancer |
| NCT02620423 | Reolysin | Pembrolizumab | PD-1 | I | Pancreatic Adenocarcinoma |
| NCT04445844 | Pelareorep | Retifanlimab | PD-1 | II | Breast cancers |
| NCT04215146 | Pelareorep | Avelumab | PD-L1 | II | Metastatic breast cancer |
| Trial | Virus Type | OV Agent | ICB Therapy | Phase (s) | Cancer (s) | |
|---|---|---|---|---|---|---|
| Agent | Target | |||||
| NCT03004183 | Adenovirus | ADV/HSVtk | Pembrolizumab | PD-1 | II | Metastatic NSCLC, Metastatic TNBC |
| NCT02798406 | Adenovirus | DNX-2401 | Pembrolizumab | PD-1 | II | Glioblastoma, Gliosarcoma |
| NCT03003676 | Adenovirus | ONCOS-102 | Pembrolizumab | PD-1 | I | Advanced/unresectable melanoma progressing after PD-1 blockade |
| NCT03408587 | Coxsackie | CAVATAK | Ipilimumab | CTLA-4 | Ib | Uveal Melanoma with Liver Metastases |
| NCT02565992 | Coxsackie | CAVATAK | Pembrolizumab | PD-1 | I | Advanced Melanoma |
| NCT02824965 | Coxsackie | CAVATAK | Pembrolizumab | PD-1 | I, II | Advanced NSCLC |
| NCT03153085 | HSV | HF10 (TBI-1401) | Ipilimumab | CTLA-4 | II | Unresectable/Metastatic Melanoma |
| NCT02272855 | HSV | HF10 (TBI-1401) | Ipilimumab | CTLA-4 | II | Unresectable/Metastatic Melanoma |
| NCT03259425 | HSV | HF10 (TBI-1401) | Nivolumab | PD-1 | II | Resectable Stage IIIB/C, IV Melanoma |
| NCT01740297 | HSV | T-Vec | Ipilimumab | CTLA-4 | Ib, II | Unresected Stage IIIb/IV melanoma |
| NCT02263508 | HSV | T-Vec | Pembrolizumab | PD-1 | Ib, III | Unresectable Stage IIIb/IV Melanoma |
| NCT02626000 | HSV | T-Vec | Pembrolizumab | PD-1 | Ib, III | Recurrent/Metastatic HNSCC |
| NCT02879760 | Maraba Virus | MG1-MAGEA3 | Pembrolizumab | PD-1 | I, II | Previously treated NSCLC |
| NCT03206073 | Vaccinia | Pexa Vec | Durvalumab | PD-L1 | I, II | Refractory Colorectal Cancer |
| Tremelimumab | CTLA-4 | |||||
| NCT02977156 | Vaccinia | Pexa Vec | Ipilimumab | CTLA-4 | I | Metastatic/Advanced Solid Tumors |
| NCT03071094 | Vaccinia | Pexa Vec | Nivolumab | PD-1 | I, IIa | Advanced HCC |
| NCT04185311 | HSV | T-Vec | Ipilimumab | CTLA-4 | I | Localized Breast Cancer |
| Nivolumab | PD-1 | |||||
| NCT03889275 | Newcastle disease virus | MEDI5395 | Durvalumab | PD-L1 | I | Advanced Solid Tumors |
| NCT04301011 | Vaccinia | TBio-6517 | Pembrolizumab | PD-1 | I, II | Triple Negative Breast Cancer Microsatellite Stable Colorectal Cancer |
| NCT04735978 | HSV | RP3 | Anti-PD-1 mAb | PD-1 | I | Advanced Solid Tumor |
| NCT04348916 | HSV | ONCR-177 | Pembrolizumab | PD-1 | I | Advanced Solid Tumors |
| NCT03294083 | Vaccinia | Pexa Vec | Cemiplimab | PD-1 | I, II | Renal Cell Carcinoma |
| NCT04755543 | HSV | OH2 | LP002 | PD-1 | I | Digestive System Neoplasms |
| NCT04386967 | HSV | OH2 | Keytruda | PD-1 | I, II | Solid Tumors, Melanoma |
| NCT04616443 | HSV | OH2 | HX008 | PD-1 | I, II | Melanoma |
| NCT03866525 | HSV | OH2 | HX008 | PD-1 | I, II | Solid Tumors, Gastrointestinal Cancer |
| NCT03206073 | Vaccinia | Pexa-Vec | Durvalumab | PD-L1 | I, II | Colorectal Neoplasms |
| Tremelimumab | CTLA-4 | |||||
| NCT04665362 | Alphavirus | M1 | SHR-1210 | PD-1 | I | Advanced/Metastatic Hepatocellular Carcinoma |
| NCT04685499 | Adenovirus | OBP-301 | Pembrolizumab | PD-1 | II | HNSCC |
| Trial | TAM-Directed Agent | ICB Therapy | Phase (S) | Cancer (S) | ||
|---|---|---|---|---|---|---|
| Agent | Target | Agent | Target | |||
| NCT02323191 | Emactuzumab | CSF1R | Atezolizumab | PD-L1 | I | Locally advanced or metastatic solid tumors |
| NCT02880371 | ARRY-382 | CSF1R | Pembrolizumab | PD-1 | I/II | Advanced solid tumors |
| NCT02777710 | Pexidartinib | CSF1R | Durvalumab | PD-L1 | I | Colorectal cancer; Pancreatic cancer; Metastatic cancer; Advanced cancer |
| NCT03238027 | SNFX-6352 | CSF1R | Durvalumab | PD-L1 | I | Solid tumor; Metastatic tumor; Locally advanced malignant neoplasm; Unresectable malignant neoplasm |
| NCT02829723 | BLZ945 | CSF1R | PDR001 | PD-1 | I/II | Advanced solid tumors |
| NCT03158272 | Cabiralizumab | CSF1R | Nivolumab | PD-1 | I | Advanced malignancies |
| NCT02713529 | AMG820 | CSF1R | Pembrolizumab | PD-1 | I/II | Pancreatic cancer; Colorectal cancer; Non-small cell lung cancer |
| NCT03123783 | APX005M | CD40 | Nivolumab | PD-1 | I/II | Non-small cell lung cancer; Metastatic melanoma |
| NCT02304393 | Selicrelumab | CD40 | Atezolizumab | PD-L1 | I | Solid tumors |
| NCT02637531 | IPI-549 | PI3Kγ | Nivolumab | PD-1 | I | Advanced solid tumor; non-small cell lung cancer; melanoma; breast cancer |
| NCT02890368 | TTI-621 | SIRPα | Nivolumab | PD-1 | I | Solid tumors; melanoma; merkel-cell carcinoma; squamous cell carcinoma; breast carcinoma |
| Pembrolizumab | PD-1 | |||||
| Atezolizumab | PD-L1 | |||||
| Durvalumab | PD-L1 | |||||
| NCT03530683 | TTI-621 | SIRPα | Nivolumab | PD-1 | I | Lymphoma; myeloma |
| Pembrolizumab | PD-1 | |||||
| NCT03681951 | GSK3145095 | RIP | Pembrolizumab | PD-1 | I/II | Neoplasms; pancreatic |
| NCT03435640 | NKTR262 | TLR7/8 | Nivolumab | PD-1 | I/II | Melanoma; merkel cell carcinoma; breast cancer; renal cell carcinoma; colorectal cancer |
| NCT02880371 | ARRY-382 | CSF1R | Pembrolizumab | PD-1 | II | Advanced solid tumors |
| NCT03153410 | IMC-CS4 | CSF1R | Pembrolizumab | PD-1 | I | PDAC |
| NCT02526017 | FPA008 (Cabiralizumab) | CSF1R | Nivolumab | PD-1 | I | Advanced solid tumors |
| NCT03708224 | Emactuzumab | CSF1R | Atezolizumab | PD-L1 | II | Advanced HNSCC |
| NCT03184870 | BMS-813160 | CCR2 | Nivolumab | PD-1 | I/II | PDAC, CRC |
| NCT03496662 | I/II | PDAC | ||||
| NCT03767582 | I/II | Locally advanced PDAC | ||||
| NCT03447314 | GSK1795091 | TLR4 | Pembrolizumab | PD-1 | I | Advanced solid tumors |
| NCT03445533 | IMO-2125 | TLR7/8 | Ipilimumab | CTLA-4 | III | Metastatic melanoma |
| NCT02644967 | IMO-2125 | TLR7/8 | Pembrolizumab | PD-1 | I/II | Metastatic melanoma |
| NCT02521870 | SD101 | TLR9 | Pembrolizumab | PD-1 | Ib/II | Metastatic melanoma, recurrent HNSCC |
| NCT03007732 | I/II | Solid tumors | ||||
| NCT03618641 | CMP-001 | TLR9 | Nivolumab | PD-1 | II | Melanoma |
| NCT03507699 | Ipilimumab | CTLA-4 | ||||
| NCT02403271 | Ibrutinib | BTK | Durvalumab | PD-L1 | I/II | Relapsed or refractory solid tumors |
| NCT02376699 | SEA-CD40 | CD40 | Pembrolizumab | PD-1 | I | Solid tumors |
| NCT01103635 | CP-870, 893 | CD40 | Tremelimumab | CTLA-4 | I | Metastatic melanoma |
| NCT02760797 | R07009879 (Selicrelumab) | CD40 | Anti-PD-L1 | PD-L1 | I | Advanced solid tumors |
| NCT02665416 | Bevacizumab/Vanucizumab | VEGF-A | I | Advanced solid tumors | ||
| NCT02953782 | Hu5F9-G4 | CD47 | Cetuximab | EGFR | I | Advanced solid malignancies and colorectal carcinoma |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, V.; Giacomantonio, M.A.; Gujar, S. Role of Myeloid Cells in Oncolytic Reovirus-Based Cancer Therapy. Viruses 2021, 13, 654. https://doi.org/10.3390/v13040654
Kumar V, Giacomantonio MA, Gujar S. Role of Myeloid Cells in Oncolytic Reovirus-Based Cancer Therapy. Viruses. 2021; 13(4):654. https://doi.org/10.3390/v13040654
Chicago/Turabian StyleKumar, Vishnupriyan, Michael A. Giacomantonio, and Shashi Gujar. 2021. "Role of Myeloid Cells in Oncolytic Reovirus-Based Cancer Therapy" Viruses 13, no. 4: 654. https://doi.org/10.3390/v13040654
APA StyleKumar, V., Giacomantonio, M. A., & Gujar, S. (2021). Role of Myeloid Cells in Oncolytic Reovirus-Based Cancer Therapy. Viruses, 13(4), 654. https://doi.org/10.3390/v13040654