
The Role of the Host Ubiquitin System in Promoting Replication of Emergent Viruses


Abstract
1. Introduction
2. Ubiquitin System and Virus Entry
3. The Ubiquitin System in Promoting Virus Replication
4. The Ubiquitin System in Virus Assembly and Budding
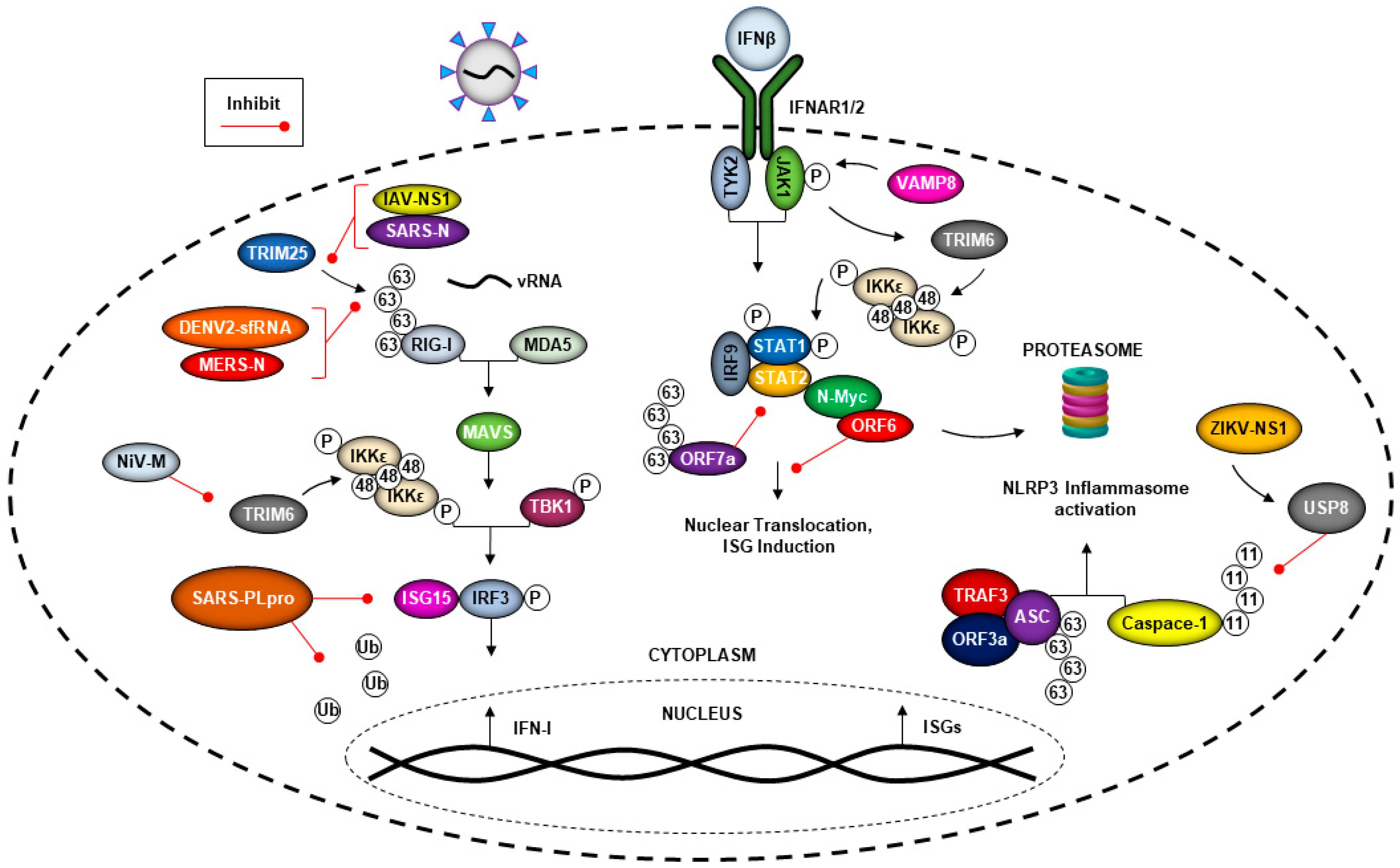
5. The Ubiquitin System and Viral Evasion of the Type-I Interferon (IFN-I) Response
5.1. Coronaviruses Can Evade the Immune Response by Hijacking the Ubiquitin System
5.2. Flavivirus Manipulation of the Ubiquitin System and the IFN Response
5.3. Nipah Virus Matrix Protein (NiV-M) Inhibits TRIM6-Mediated IFN Responses
6. Conclusions and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Li, X.; Elmira, E.; Rohondia, S.; Wang, J.; Liu, J.; Dou, Q.P. A patent review of the ubiquitin ligase system: 2015–2018. Expert. Opin. Ther. Pat. 2018, 28, 919–937. [Google Scholar] [CrossRef]
- Zhuang, J.; Shirazi, F.; Singh, R.K.; Kuiatse, I.; Wang, H.; Lee, H.C.; Berkova, Z.; Berger, A.; Hyer, M.; Chattopadhyay, N.; et al. Ubiquitin-activating enzyme inhibition induces an unfolded protein response and overcomes drug resistance in myeloma. Blood 2019, 133, 1572–1584. [Google Scholar] [CrossRef]
- Ye, Y.; Rape, M. Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 2009, 10, 755–764. [Google Scholar] [CrossRef]
- Hage, A.; Rajsbaum, R. To TRIM or not to TRIM: The balance of host-virus interactions mediated by the ubiquitin system. J. Gen. Virol. 2019, 100, 1641–1662. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Hage, A.; Van Tol, S.; Rajsbaum, R. TRIM Proteins in Host Defense and Viral Pathogenesis. Curr. Clin. Microbiol. Rep. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Liu, H.; Zu, X.; Liu, Y.; Chen, L.; Zhu, X.; Zhang, L.; Zhou, Z.; Xiao, G.; Wang, W. The ubiquitin-proteasome system is essential for the productive entry of Japanese encephalitis virus. Virology 2016, 498, 116–127. [Google Scholar] [CrossRef] [PubMed]
- Byk, L.A.; Iglesias, N.G.; De Maio, F.A.; Gebhard, L.G.; Rossi, M.; Gamarnik, A.V. Dengue Virus Genome Uncoating Requires Ubiquitination. mBio 2016, 7. [Google Scholar] [CrossRef]
- Dejarnac, O.; Hafirassou, M.L.; Chazal, M.; Versapuech, M.; Gaillard, J.; Perera-Lecoin, M.; Umana-Diaz, C.; Bonnet-Madin, L.; Carnec, X.; Tinevez, J.Y.; et al. TIM-1 Ubiquitination Mediates Dengue Virus Entry. Cell Rep. 2018, 23, 1779–1793. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Vargas-Cuartas, O.; Gallego-Gomez, J.C.; Shi, P.Y.; Padilla-Sanabria, L.; Castaño-Osorio, J.C.; Rajsbaum, R. K48-linked polyubiquitination of dengue virus NS1 protein inhibits its interaction with the viral partner NS4B. Virus Res. 2018, 246, 1–11. [Google Scholar] [CrossRef]
- Ramanathan, H.N.; Zhang, S.; Douam, F.; Mar, K.B.; Chang, J.; Yang, P.L.; Schoggins, J.W.; Ploss, A.; Lindenbach, B.D. A Sensitive Yellow Fever Virus Entry Reporter Identifies Valosin-Containing Protein (VCP/p97) as an Essential Host Factor for Flavivirus Uncoating. mBio 2020, 11. [Google Scholar] [CrossRef]
- Giraldo, M.I.; Xia, H.; Aguilera-Aguirre, L.; Hage, A.; Van Tol, S.; Shan, C.; Xie, X.; Sturdevant, G.L.; Robertson, S.J.; McNally, K.L.; et al. Envelope protein ubiquitination drives entry and pathogenesis of Zika virus. Nature 2020, 585, 414–419. [Google Scholar] [CrossRef]
- Su, W.C.; Chen, Y.C.; Tseng, C.H.; Hsu, P.W.; Tung, K.F.; Jeng, K.S.; Lai, M.M. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc. Natl. Acad. Sci. USA 2013, 110, 17516–17521. [Google Scholar] [CrossRef]
- Rudnicka, A.; Yamauchi, Y. Ubiquitin in Influenza Virus Entry and Innate Immunity. Viruses 2016, 8, 293. [Google Scholar] [CrossRef]
- Su, W.C.; Yu, W.Y.; Huang, S.H.; Lai, M.M.C. Ubiquitination of the Cytoplasmic Domain of Influenza A Virus M2 Protein Is Crucial for Production of Infectious Virus Particles. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Bauer, M.; Flatt, J.W.; Seiler, D.; Cardel, B.; Emmenlauer, M.; Boucke, K.; Suomalainen, M.; Hemmi, S.; Greber, U.F. The E3 Ubiquitin Ligase Mind Bomb 1 Controls Adenovirus Genome Release at the Nuclear Pore Complex. Cell Rep. 2019, 29, 3785–3795. [Google Scholar] [CrossRef]
- Bharaj, P.; Atkins, C.; Luthra, P.; Giraldo, M.I.; Dawes, B.E.; Miorin, L.; Johnson, J.R.; Krogan, N.J.; Basler, C.F.; Freiberg, A.N.; et al. The Host E3-Ubiquitin Ligase TRIM6 Ubiquitinates the Ebola Virus VP35 Protein and Promotes Virus Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Sagum, C.A.; Bedford, M.T.; Sidhu, S.S.; Sudol, M.; Harty, R.N. ITCH E3 Ubiquitin Ligase Interacts with Ebola Virus VP40 To Regulate Budding. J. Virol. 2016, 90, 9163–9171. [Google Scholar] [CrossRef]
- Baz-Martinez, M.; El Motiam, A.; Ruibal, P.; Condezo, G.N.; De la Cruz-Herrera, C.F.; Lang, V.; Collado, M.; San Martin, C.; Rodriguez, M.S.; Munoz-Fontela, C.; et al. Regulation of Ebola virus VP40 matrix protein by SUMO. Sci. Rep. 2016, 6, 37258. [Google Scholar] [CrossRef]
- Yasuda, J.; Nakao, M.; Kawaoka, Y.; Shida, H. Nedd4 regulates egress of Ebola virus-like particles from host cells. J. Virol. 2003, 77, 9987–9992. [Google Scholar] [CrossRef]
- Ziegler, C.M.; Dang, L.; Eisenhauer, P.; Kelly, J.A.; King, B.R.; Klaus, J.P.; Manuelyan, I.; Mattice, E.B.; Shirley, D.J.; Weir, M.E.; et al. NEDD4 family ubiquitin ligases associate with LCMV Z’s PPXY domain and are required for virus budding, but not via direct ubiquitination of Z. PLoS Pathog. 2019, 15. [Google Scholar] [CrossRef]
- Pentecost, M.; Vashisht, A.A.; Lester, T.; Voros, T.; Beaty, S.M.; Park, A.; Wang, Y.E.; Yun, T.E.; Freiberg, A.N.; Wohlschlegel, J.A.; et al. Evidence for ubiquitin-regulated nuclear and subnuclear trafficking among Paramyxovirinae matrix proteins. PLoS Pathog. 2015, 11, 1004739. [Google Scholar] [CrossRef]
- Bharaj, P.; Wang, Y.E.; Dawes, B.E.; Yun, T.E.; Park, A.; Yen, B.; Basler, C.F.; Freiberg, A.N.; Lee, B.; Rajsbaum, R. The Matrix Protein of Nipah Virus Targets the E3-Ubiquitin Ligase TRIM6 to Inhibit the IKKepsilon Kinase-Mediated Type-I IFN Antiviral Response. PLoS Pathog. 2016, 12, 1005880. [Google Scholar] [CrossRef]
- Cheng, W.; Chen, S.; Li, R.; Chen, Y.; Wang, M.; Guo, D. Severe acute respiratory syndrome coronavirus protein 6 mediates ubiquitin-dependent proteosomal degradation of N-Myc (and STAT) interactor. Virol. Sin. 2015, 30, 153–161. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234. [Google Scholar] [CrossRef]
- Sanchez-Aparicio, M.T.; Feinman, L.J.; Garcia-Sastre, A.; Shaw, M.L. Paramyxovirus V Proteins Interact with the RIG-I/TRIM25 Regulatory Complex and Inhibit RIG-I Signaling. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Siu, K.L.; Yuen, K.S.; Castaño-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]
- Marsh, M.; Helenius, A. Virus entry: Open sesame. Cell 2006, 124, 729–740. [Google Scholar] [CrossRef]
- Cohen, F.S. How Viruses Invade Cells. Biophys. J. 2016, 110, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Mohanram, V.; Simonson, O.E.; Smith, C.I.; Spetz, A.L.; Mohamed, A.J. Proteasome inhibitors block HIV-1 replication by affecting both cellular and viral targets. Biochem. Biophys. Res. Commun. 2009, 385, 100–105. [Google Scholar] [CrossRef]
- Casorla-Pérez, L.A.; López, T.; López, S.; Arias, C.F. The Ubiquitin-Proteasome System Is Necessary for Efficient Replication of Human Astrovirus. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Schneider, S.M.; Pritchard, S.M.; Wudiri, G.A.; Trammell, C.E.; Nicola, A.V. Early Steps in Herpes Simplex Virus Infection Blocked by a Proteasome Inhibitor. mBio 2019, 10. [Google Scholar] [CrossRef]
- Schneider, M.; Ackermann, K.; Stuart, M.; Wex, C.; Protzer, U.; Schätzl, H.M.; Gilch, S. Severe acute respiratory syndrome coronavirus replication is severely impaired by MG132 due to proteasome-independent inhibition of M-calpain. J. Virol. 2012, 86, 10112–10122. [Google Scholar] [CrossRef]
- Schneider, S.M.; Lee, B.H.; Nicola, A.V. Viral entry and the ubiquitin-proteasome system. Cell. Microbiol. 2020. [Google Scholar] [CrossRef]
- Yu, G.Y.; Lai, M.M. The ubiquitin-proteasome system facilitates the transfer of murine coronavirus from endosome to cytoplasm during virus entry. J. Virol. 2005, 79, 644–648. [Google Scholar] [CrossRef]
- Van den Boom, J.; Meyer, H. VCP/p97-Mediated Unfolding as a Principle in Protein Homeostasis and Signaling. Mol. Cell 2018, 69, 182–194. [Google Scholar] [CrossRef]
- Ye, Y.; Tang, W.K.; Zhang, T.; Xia, D. A Mighty "Protein Extractor" of the Cell: Structure and Function of the p97/CDC48 ATPase. Front. Mol. Biosci. 2017, 4, 39. [Google Scholar] [CrossRef]
- Sluimer, J.; Distel, B. Regulating the human HECT E3 ligases. Cell Mol. Life Sci. 2018, 75, 3121–3141. [Google Scholar] [CrossRef]
- Banerjee, I.; Miyake, Y.; Nobs, S.P.; Schneider, C.; Horvath, P.; Kopf, M.; Matthias, P.; Helenius, A.; Yamauchi, Y. Influenza A virus uses the aggresome processing machinery for host cell entry. Science 2014, 346, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Hao, R.; Nanduri, P.; Rao, Y.; Panichelli, R.S.; Ito, A.; Yoshida, M.; Yao, T.P. Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 2013, 51, 819–828. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Kovacs, J.J.; McLaurin, A.; Vance, J.M.; Ito, A.; Yao, T.P. The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 2003, 115, 727–738. [Google Scholar] [CrossRef]
- Rajsbaum, R.; García-Sastre, A. Virology. Unanchored ubiquitin in virus uncoating. Science 2014, 346, 427–428. [Google Scholar] [CrossRef]
- Chen, Y.; Lear, T.; Evankovich, J.; Larsen, M.; Lin, B.; Alfaras, I.; Kennerdell, J.; Salminen, L.; Camarco, D.; Lockwood, K.; et al. A high throughput screen for TMPRSS2 expression identifies FDA-approved and clinically advanced compounds that can limit SARS-CoV-2 entry. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Iwata-Yoshikawa, N.; Okamura, T.; Shimizu, Y.; Hasegawa, H.; Takeda, M.; Nagata, N. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Longhitano, L.; Tibullo, D.; Giallongo, C.; Lazzarino, G.; Tartaglia, N.; Galimberti, S.; Li Volti, G.; Palumbo, G.A.; Liso, A. Proteasome Inhibitors as a Possible Therapy for SARS-CoV-2. Int. J. Mol. Sci. 2020, 21, 3622. [Google Scholar] [CrossRef]
- Rodriguez, A.; Pérez-González, A.; Nieto, A. Influenza virus infection causes specific degradation of the largest subunit of cellular RNA polymerase II. J. Virol. 2007, 81, 5315–5324. [Google Scholar] [CrossRef] [PubMed]
- Liao, T.L.; Wu, C.Y.; Su, W.C.; Jeng, K.S.; Lai, M.M. Ubiquitination and deubiquitination of NP protein regulates influenza A virus RNA replication. EMBO J. 2010, 29, 3879–3890. [Google Scholar] [CrossRef]
- Kirui, J.; Mondal, A.; Mehle, A. Ubiquitination Upregulates Influenza Virus Polymerase Function. J. Virol. 2016, 90, 10906–10914. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.C.; Jeng, K.S.; Lai, M.M.C. CNOT4-Mediated Ubiquitination of Influenza A Virus Nucleoprotein Promotes Viral RNA Replication. mBio 2017, 8. [Google Scholar] [CrossRef]
- Pinto, L.H.; Holsinger, L.J.; Lamb, R.A. Influenza virus M2 protein has ion channel activity. Cell 1992, 69, 517–528. [Google Scholar] [CrossRef]
- Rossman, J.S.; Jing, X.; Leser, G.P.; Lamb, R.A. Influenza virus M2 protein mediates ESCRT-independent membrane scission. Cell 2010, 142, 902–913. [Google Scholar] [CrossRef]
- Stott, R.J.; Strecker, T.; Foster, T.L. Distinct Molecular Mechanisms of Host Immune Response Modulation by Arenavirus NP and Z Proteins. Viruses 2020, 12, 784. [Google Scholar] [CrossRef]
- Baillet, N.; Krieger, S.; Carnec, X.; Mateo, M.; Journeaux, A.; Merabet, O.; Caro, V.; Tangy, F.; Vidalain, P.O.; Baize, S. E3 Ligase ITCH Interacts with the Z Matrix Protein of Lassa and Mopeia Viruses and Is Required for the Release of Infectious Particles. Viruses 2019, 12, 49. [Google Scholar] [CrossRef]
- Han, Z.; Sagum, C.A.; Takizawa, F.; Ruthel, G.; Berry, C.T.; Kong, J.; Sunyer, J.O.; Freedman, B.D.; Bedford, M.T.; Sidhu, S.S.; et al. Ubiquitin Ligase WWP1 Interacts with Ebola Virus VP40 To Regulate Egress. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Urata, S.; Yasuda, J. Regulation of Marburg virus (MARV) budding by Nedd4.1: A different WW domain of Nedd4.1 is critical for binding to MARV and Ebola virus VP40. J. Gen. Virol. 2010, 91, 228–234. [Google Scholar] [CrossRef]
- Lafont, F.; Simons, K. Raft-partitioning of the ubiquitin ligases Cbl and Nedd4 upon IgE-triggered cell signaling. Proc. Natl. Acad. Sci. USA 2001, 98, 3180–3184. [Google Scholar] [CrossRef]
- Harvey, K.F.; Kumar, S. Nedd4-like proteins: An emerging family of ubiquitin-protein ligases implicated in diverse cellular functions. Trends Cell Biol. 1999, 9, 166–169. [Google Scholar] [CrossRef]
- Bonthius, D.J.; Perlman, S. Congenital viral infections of the brain: Lessons learned from lymphocytic choriomeningitis virus in the neonatal rat. PLoS Pathog. 2007, 3, 149. [Google Scholar] [CrossRef]
- Wang, Y.E.; Park, A.; Lake, M.; Pentecost, M.; Torres, B.; Yun, T.E.; Wolf, M.C.; Holbrook, M.R.; Freiberg, A.N.; Lee, B. Ubiquitin-regulated nuclear-cytoplasmic trafficking of the Nipah virus matrix protein is important for viral budding. PLoS Pathog. 2010, 6, 1001186. [Google Scholar] [CrossRef]
- Hoffmann, J.; Akira, S. Innate immunity. Curr. Opin. Immunol. 2013, 25, 1–3. [Google Scholar] [CrossRef]
- Oshiumi, H.; Miyashita, M.; Inoue, N.; Okabe, M.; Matsumoto, M.; Seya, T. The ubiquitin ligase Riplet is essential for RIG-I-dependent innate immune responses to RNA virus infection. Cell Host Microbe 2010, 8, 496–509. [Google Scholar] [CrossRef]
- Oshiumi, H. Recent Advances and Contradictions in the Study of the Individual Roles of Ubiquitin Ligases That Regulate RIG-I-Like Receptor-Mediated Antiviral Innate Immune Responses. Front. Immunol. 2020, 11, 1296. [Google Scholar] [CrossRef]
- Liu, B.; Gao, C. Regulation of MAVS activation through post-translational modifications. Curr. Opin. Immunol. 2018, 50, 75–81. [Google Scholar] [CrossRef]
- Iwasaki, A. A virological view of innate immune recognition. Annu. Rev. Microbiol. 2012, 66, 177–196. [Google Scholar] [CrossRef] [PubMed]
- Van Tol, S.; Atkins, C.; Bharaj, P.; Johnson, K.N.; Hage, A.; Freiberg, A.N.; Rajsbaum, R. VAMP8 Contributes to the TRIM6-Mediated Type I Interferon Antiviral Response during West Nile Virus Infection. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Totura, A.L.; Baric, R.S. SARS coronavirus pathogenesis: Host innate immune responses and viral antagonism of interferon. Curr. Opin. Virol. 2012, 2, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Frieman, M.; Ratia, K.; Johnston, R.E.; Mesecar, A.D.; Baric, R.S. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 2009, 83, 6689–6705. [Google Scholar] [CrossRef]
- Hu, Y.; Li, W.; Gao, T.; Cui, Y.; Jin, Y.; Li, P.; Ma, Q.; Liu, X.; Cao, C. The Severe Acute Respiratory Syndrome Coronavirus Nucleocapsid Inhibits Type I Interferon Production by Interfering with TRIM25-Mediated RIG-I Ubiquitination. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Niemeyer, D.; Mosbauer, K.; Klein, E.M.; Sieberg, A.; Mettelman, R.C.; Mielech, A.M.; Dijkman, R.; Baker, S.C.; Drosten, C.; Muller, M.A. The papain-like protease determines a virulence trait that varies among members of the SARS-coronavirus species. PLoS Pathog. 2018, 14, 1007296. [Google Scholar] [CrossRef]
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2020. [Google Scholar] [CrossRef]
- Cao, Z.; Xia, H.; Rajsbaum, R.; Xia, X.; Wang, H.; Shi, P.Y. Ubiquitination of SARS-CoV-2 ORF7a promotes antagonism of interferon response. Cell Mol. Immunol. 2021. [Google Scholar] [CrossRef]
- Gack, M.U.; Shin, Y.C.; Joo, C.H.; Urano, T.; Liang, C.; Sun, L.; Takeuchi, O.; Akira, S.; Chen, Z.; Inoue, S.; et al. TRIM25 RING-finger E3 ubiquitin ligase is essential for RIG-I-mediated antiviral activity. Nature 2007, 446, 916–920. [Google Scholar] [CrossRef]
- Gack, M.U.; Albrecht, R.A.; Urano, T.; Inn, K.S.; Huang, I.C.; Carnero, E.; Farzan, M.; Inoue, S.; Jung, J.U.; Garcia-Sastre, A. Influenza A virus NS1 targets the ubiquitin ligase TRIM25 to evade recognition by the host viral RNA sensor RIG-I. Cell Host Microbe 2009, 5, 439–449. [Google Scholar] [CrossRef]
- Manokaran, G.; Finol, E.; Wang, C.; Gunaratne, J.; Bahl, J.; Ong, E.Z.; Tan, H.C.; Sessions, O.M.; Ward, A.M.; Gubler, D.J.; et al. Dengue subgenomic RNA binds TRIM25 to inhibit interferon expression for epidemiological fitness. Science 2015, 350, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Barretto, N.; Jukneliene, D.; Ratia, K.; Chen, Z.; Mesecar, A.D.; Baker, S.C. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 2005, 79, 15189–15198. [Google Scholar] [CrossRef]
- Lindner, H.A.; Fotouhi-Ardakani, N.; Lytvyn, V.; Lachance, P.; Sulea, T.; Menard, R. The papain-like protease from the severe acute respiratory syndrome coronavirus is a deubiquitinating enzyme. J. Virol. 2005, 79, 15199–15208. [Google Scholar] [CrossRef]
- Freitas, B.T.; Scholte, F.E.M.; Bergeron, E.; Pegan, S.D. How ISG15 combats viral infection. Virus Res. 2020, 286, 198036. [Google Scholar] [CrossRef]
- Devaraj, S.G.; Wang, N.; Chen, Z.; Chen, Z.; Tseng, M.; Barretto, N.; Lin, R.; Peters, C.J.; Tseng, C.T.; Baker, S.C.; et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J. Biol. Chem. 2007, 282, 32208–32221. [Google Scholar] [CrossRef]
- Bailey-Elkin, B.A.; Knaap, R.C.; Johnson, G.G.; Dalebout, T.J.; Ninaber, D.K.; Van Kasteren, P.B.; Bredenbeek, P.J.; Snijder, E.J.; Kikkert, M.; Mark, B.L. Crystal structure of the Middle East respiratory syndrome coronavirus (MERS-CoV) papain-like protease bound to ubiquitin facilitates targeted disruption of deubiquitinating activity to demonstrate its role in innate immune suppression. J. Biol. Chem. 2014, 289, 34667–34682. [Google Scholar] [CrossRef]
- Shin, D.; Mukherjee, R.; Grewe, D.; Bojkova, D.; Baek, K.; Bhattacharya, A.; Schulz, L.; Widera, M.; Mehdipour, A.R.; Tascher, G.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. [Google Scholar] [CrossRef]
- Freitas, B.T.; Durie, I.A.; Murray, J.; Longo, J.E.; Miller, H.C.; Crich, D.; Hogan, R.J.; Tripp, R.A.; Pegan, S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. [Google Scholar] [CrossRef]
- Klemm, T.; Ebert, G.; Calleja, D.J.; Allison, C.C.; Richardson, L.W.; Bernardini, J.P.; Lu, B.G.; Kuchel, N.W.; Grohmann, C.; Shibata, Y.; et al. Mechanism and inhibition of the papain-like protease, PLpro, of SARS-CoV-2. EMBO J. 2020, 39, 106275. [Google Scholar] [CrossRef]
- Zheng, Y.; Liu, Q.; Wu, Y.; Ma, L.; Zhang, Z.; Liu, T.; Jin, S.; She, Y.; Li, Y.P.; Cui, J. Zika virus elicits inflammation to evade antiviral response by cleaving cGAS via NS1-caspase-1 axis. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Aguirre, S.; Luthra, P.; Sanchez-Aparicio, M.T.; Maestre, A.M.; Patel, J.; Lamothe, F.; Fredericks, A.C.; Tripathi, S.; Zhu, T.; Pintado-Silva, J.; et al. Dengue virus NS2B protein targets cGAS for degradation and prevents mitochondrial DNA sensing during infection. Nat. Microbiol. 2017, 2, 17037. [Google Scholar] [CrossRef]
- Ding, Q.; Gaska, J.M.; Douam, F.; Wei, L.; Kim, D.; Balev, M.; Heller, B.; Ploss, A. Species-specific disruption of STING-dependent antiviral cellular defenses by the Zika virus NS2B3 protease. Proc. Natl. Acad. Sci. USA 2018, 115, E6310–E6318. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, S.; Maestre, A.M.; Pagni, S.; Patel, J.R.; Savage, T.; Gutman, D.; Maringer, K.; Bernal-Rubio, D.; Shabman, R.S.; Simon, V.; et al. DENV inhibits type I IFN production in infected cells by cleaving human STING. PLoS Pathog. 2012, 8, 1002934. [Google Scholar] [CrossRef] [PubMed]
- Van Tol, S.; Hage, A.; Giraldo, M.I.; Bharaj, P.; Rajsbaum, R. The TRIMendous Role of TRIMs in Virus-Host Interactions. Vaccines 2017, 5, 23. [Google Scholar] [CrossRef]
- Laurent-Rolle, M.; Morrison, J. The Role of NS5 Protein in Determination of Host Cell Range for Yellow Fever Virus. DNA Cell Biol. 2019, 38, 1414–1417. [Google Scholar] [CrossRef]
- Miorin, L.; Laurent-Rolle, M.; Pisanelli, G.; Co, P.H.; Albrecht, R.A.; Garcia-Sastre, A.; Morrison, J. Host-Specific NS5 Ubiquitination Determines Yellow Fever Virus Tropism. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Ashour, J.; Laurent-Rolle, M.; Shi, P.Y.; Garcia-Sastre, A. NS5 of dengue virus mediates STAT2 binding and degradation. J. Virol. 2009, 83, 5408–5418. [Google Scholar] [CrossRef]
- Morrison, J.; Laurent-Rolle, M.; Maestre, A.M.; Rajsbaum, R.; Pisanelli, G.; Simon, V.; Mulder, L.C.; Fernandez-Sesma, A.; Garcia-Sastre, A. Dengue virus co-opts UBR4 to degrade STAT2 and antagonize type I interferon signaling. PLoS Pathog. 2013, 9, 1003265. [Google Scholar] [CrossRef] [PubMed]
- Grant, A.; Ponia, S.S.; Tripathi, S.; Balasubramaniam, V.; Miorin, L.; Sourisseau, M.; Schwarz, M.C.; Sanchez-Seco, M.P.; Evans, M.J.; Best, S.M.; et al. Zika Virus Targets Human STAT2 to Inhibit Type I Interferon Signaling. Cell Host Microbe 2016, 19, 882–890. [Google Scholar] [CrossRef] [PubMed]
- Laurent-Rolle, M.; Morrison, J.; Rajsbaum, R.; Macleod, J.M.L.; Pisanelli, G.; Pham, A.; Ayllon, J.; Miorin, L.; Martinez, C.; tenOever, B.R.; et al. The interferon signaling antagonist function of yellow fever virus NS5 protein is activated by type I interferon. Cell Host Microbe 2014, 16, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Rajsbaum, R.; Versteeg, G.A.; Schmid, S.; Maestre, A.M.; Belicha-Villanueva, A.; Martinez-Romero, C.; Patel, J.R.; Morrison, J.; Pisanelli, G.; Miorin, L.; et al. Unanchored K48-linked polyubiquitin synthesized by the E3-ubiquitin ligase TRIM6 stimulates the interferon-IKKepsilon kinase-mediated antiviral response. Immunity 2014, 40, 880–895. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Virus | Target | Mechanism of Action | Reference |
|---|---|---|---|
| JEV | Unknown | Viral internalization, and release of viral RNPs from endosome. | [6] |
| DENV | Genome uncoating | Host E1 UBA1 implicated in vRNA uncoating and or translation. | [7] |
| DENV | Host TIM-1 | Ubiquitination of TIM-1 at K338 and K346 increases virus internalization and is important for entry | [8] |
| DENV | NS1 protein | Ubiquitinated at NS1 K189, reduces interaction with viral NS4B | [9] |
| YFV | Genome uncoating | Post-fusion stage, viral uncoating | [10] |
| ZIKV | E protein | Ubiquitination of viral E protein at K38 drives viral entry and pathogenesis | [11] |
| IAV | M1 protein | Ubiquitinated by host ITCH ubiquitin ligase, affects release from endosome | [12,13] |
| IAV | M2 protein | Ubiquitination at K78 facilitates interaction with viral M1, which allows for efficient incorporation of vRNA into virion progeny | [14] |
| ADV | Unknown | Ubiquitination function of host Mib1 needed for viral uncoating, and genome release | [15] |
| EBOV | VP35 protein | Ubiquitinated at K309, enhances polymerase activity | [16] |
| EBOV | VP40 protein | VP40 interacts with ITCH and regulates the budding of EBOV virus-like particles (VLPs) | [17] |
| EBOV | VP40 protein | SUMOylated regulates the stability of VP40 | [18] |
| EBOV | Unknown | Nedd4 monoubiquitination may modulate budding of VLPs | [19] |
| LCMV | Viral Z protein | Mechanism for recruitment of ESCRT, an important complex for viral budding | [20] |
| NiV, HeV, SeV, Mumps | Matrix protein | Nuclear–Cytoplasmic trafficking of the matrix protein. | [21] |
| NiV | M protein | Viral M protein prevents unanchored K48 Ubiquitin activation of IKKε through proteasome-independent TRIM6 degradation | [22] |
| SARS-CoV | ORF6 protein | ORF6 can interact with N-myc and STAT interactor, leading protein degradation through the ubiquitin proteasome pathway | [23] |
| SARS-CoV-2 | ORF7a protein | K63 polyubiquitination on K119 suppresses STAT2 phosphorylation | [24] |
| SARS-CoV, MERS-CoV | Interference of RIG-I ubiquitination | Viral N protein can interact and block TRIM25 from ubiquitinating RIG-1 | [25] |
| SARS-CoV | Host ASC | ORF3a, TRAF and ASC association results in K63 polyubiquitination of ASC and NLRP3 inflammasome activation | [26] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Valerdi, K.M.; Hage, A.; van Tol, S.; Rajsbaum, R.; Giraldo, M.I. The Role of the Host Ubiquitin System in Promoting Replication of Emergent Viruses. Viruses 2021, 13, 369. https://doi.org/10.3390/v13030369
Valerdi KM, Hage A, van Tol S, Rajsbaum R, Giraldo MI. The Role of the Host Ubiquitin System in Promoting Replication of Emergent Viruses. Viruses. 2021; 13(3):369. https://doi.org/10.3390/v13030369
Chicago/Turabian StyleValerdi, Karl M., Adam Hage, Sarah van Tol, Ricardo Rajsbaum, and Maria I. Giraldo. 2021. "The Role of the Host Ubiquitin System in Promoting Replication of Emergent Viruses" Viruses 13, no. 3: 369. https://doi.org/10.3390/v13030369
APA StyleValerdi, K. M., Hage, A., van Tol, S., Rajsbaum, R., & Giraldo, M. I. (2021). The Role of the Host Ubiquitin System in Promoting Replication of Emergent Viruses. Viruses, 13(3), 369. https://doi.org/10.3390/v13030369