
High Diversity of Human Non-Polio Enterovirus Serotypes Identified in Contaminated Water in Nigeria

 , ,
, ,  ,
,
Abstract
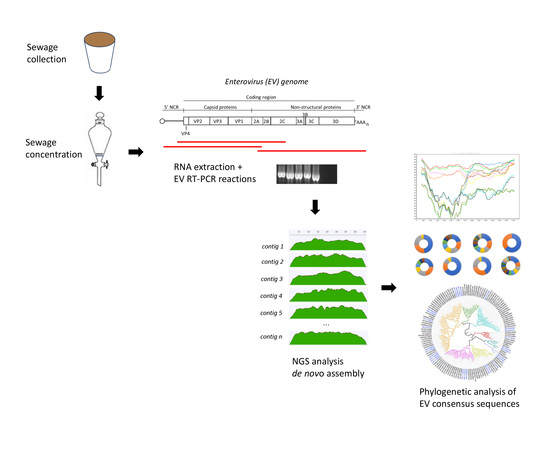
1. Introduction
2. Materials and Methods
2.1. Water Sample Collection and Processing
2.2. Pan-EV Whole Capsid-coding Region RT-PCR Amplification
- Primers 5′NCR (5′-TGGCGGAACCGACTACTTTGGGTG-3′) and CRE-R (5′-TCAATACGGTGTTTGCTCTTGAACTG-3′)
- Primers MM_EV_F2 (5′-CAGCGGAACCGACTACTTT-3′) and MM_EV_R1 (5′-AATACGGCATTTGGACTTGAACTGT-3′)
2.3. Generation of Sequencing Libraries and Quality Trimming of NGS Reads
2.4. Generation of Whole-Capsid EV Sequence Contigs by de Novo Assembly of Filtered NGS Reads
2.5. Phylogenetic Analysis of EV Strains
2.6. Whole-Genome Amplification and Sequencing of EV-D111 Strain from KAT-17-1263 Water Sample
2.7. Whole-Genome Amplification and Sequencing of EV-D94 Isolate from the Republic of Niger
2.8. Recombination Analysis
3. Results
3.1. Identification by NGS of EV Strains Present in Water Concentrates
3.2. Nucleotide Sequence Analysis of Nigerian EV Environmental Strains
3.3. Whole Genome Determination of EV-D111 in Sample KAT-17-1263
3.4. Whole Genome Determination EV-D94 14-534 Strain from the Republic of Niger
3.5. Recombination Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| EV | Enterovirus |
| E- | Echovirus |
| CV- | Coxsackievirus |
| NGS | Next Generation Sequencing |
| ES | Environmental Surveillance |
| ORF | Open Reading Frame |
| Country Abbreviations | |
| USA | United States |
| CAF | Central African Republic |
| NGR | Nigeria |
| AUS | Australia |
| MAD | Madagascar |
| NET | The Netherlands |
| IND | India |
| JPN | Japan |
| CHN | China |
| THA | Thailand |
| GER | Germany |
| HUN | Hungary |
| PNG | Papua New Guinea |
| VTM | Viet Nam |
| TKM | Turkmenistan |
| UKR | Ukraine |
| UKN | United Kingdom |
| RUS | Russia |
| LVA | Latvia |
| BAN | Bangladesh |
| KAZ | Kazakhstan |
| MAL | Malaysia |
| DNK | Denmark |
| TAJ | Tajikistan |
| GUI | Guinea |
| TUN | Tunisia |
| ETH | Ethiopia |
| PAK | Pakistan |
| IRQ | Iraq |
| NZL | New Zealand |
| GHA | Ghana |
| TWN | Taiwan |
| FIN | Finland |
| KOR | South Korea |
| FRA | France |
| HKN | Hong Kong |
| SWT | Switzerland |
| SAF | South Africa |
| SEN | Senegal |
| NIG | Niger |
| NGR | Nigeria |
| CMR | Cameroon |
| DRC | Democratic Republic of Congo |
| OMA | Oman |
| ROM | Romania |
| CHA | Chad |
| ARG | Argentina |
| CAM | Cambodia |
| MOR | Morocco |
| BEL | Belgium |
| BRA | Brazil |
| PUR | Puerto Rico |
| ROC | Republic of Congo |
| GUF | French Guiana |
| ANG | Angola |
| SWE | Sweden |
| ITA | Italy |
| MEX | Mexico |
| HON | Honduras |
References
- WHO; UNICEF. Progress on Sanitation and Drinking Water: 2015 Update and MDG Assessment; WHO: Geneva, Switzerland, 2015. [Google Scholar]
- Pallansch, M.A.; Oberste, M.S.; Whitton, L.J. Enteroviruses: Polioviruses, coxsackieviruses, echoviruses, and newer enteroviruses. In Fields’ Virology, 6th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Lulla, V.; Dinan, A.M.; Hosmillo, M.; Chaudhry, Y.; Sherry, L.; Irigoyen, N.; Nayak, K.M.; Stonehouse, N.J.; Zilbauer, M.; Goodfellow, I.; et al. An upstream protein-coding region in enteroviruses modulates virus infection in gut epithelial cells. Nat. Microbiol. 2019, 4, 280–292. [Google Scholar] [CrossRef]
- Zell, R.; Delwart, E.; Gorbalenya, A.E.; Hovi, T.; King, A.M.Q.; Knowles, N.J.; Lindberg, A.M.; Pallansch, M.A.; Palmenberg, A.C.; Reuter, G.; et al. ICTV Virus Taxonomy Profile: Picornaviridae. J. Gen. Virol. 2017, 98, 2421–2422. [Google Scholar] [CrossRef] [PubMed]
- Tapparel, C.; Siegrist, F.; Petty, T.J.; Kaiser, L. Picornavirus and enterovirus diversity with associated human diseases. Infect. Genet. Evol. 2013, 14, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Chard, A.N.; Datta, S.D.; Tallis, G.; Burns, C.C.; Wassilak, S.G.F.; Vertefeuille, J.F.; Zaffran, M. Progress Toward Polio Eradication—Worldwide, January 2018–March 2020; MMWR; CDC: Atlanta, GA, USA, 2020; Volume 69, pp. 784–789.
- Solomon, T.; Lewthwaite, P.; Perera, D.; Cardosa, M.J.; McMinn, P.; Ooi, M.H. Virology, epidemiology, pathogenesis, and control of enterovirus 71. Lancet Infect. Dis. 2010, 10, 778–790. [Google Scholar] [CrossRef]
- Messacar, K.; Asturias, E.J.; Hixon, A.M.; Van Leer-Buter, C.; Niesters, H.G.M.; Tyler, K.L.; Abzug, M.J.; Dominguez, S.R. Enterovirus D68 and acute flaccid myelitis-evaluating the evidence for causality. Lancet Infect. Dis. 2018, 18, e239–e247. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for Environmental Surveillance of Poliovirus Circulation; WHO: Geneva, Switzerland, 2003. [Google Scholar]
- Ndiaye, A.K.; Diop, P.A.; Diop, O.M. Environmental surveillance of poliovirus and non-polio enterovirus in urban sewage in Dakar, Senegal (2007–2013). Pan Afr. Med. J. 2014, 19, 243. [Google Scholar] [CrossRef]
- Apostol, L.N.; Imagawa, T.; Suzuki, A.; Masago, Y.; Lupisan, S.; Olveda, R.; Saito, M.; Omura, T.; Oshitani, H. Genetic diversity and molecular characterization of enteroviruses from sewage-polluted urban and rural rivers in the Philippines. Virus Genes 2012, 45, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Benschop, K.S.M.; van der Avoort, H.G.; Jusic, E.; Vennema, H.; van Binnendijk, R.; Duizer, E. Polio and Measles Down the Drain: Environmental Enterovirus Surveillance in the Netherlands, 2005 to 2015. Appl. Environ. Microbiol. 2017, 83, e00558-17. [Google Scholar] [CrossRef]
- Asghar, H.; Diop, O.M.; Weldegebriel, G.; Malik, F.; Shetty, S.; El Bassioni, L.; Akande, A.O.; Al Maamoun, E.; Zaidi, S.; Adeniji, A.J.; et al. Environmental surveillance for polioviruses in the Global Polio Eradication Initiative. J. Infect. Dis. 2014, 210, S294–S303. [Google Scholar] [CrossRef] [PubMed]
- Ng, T.F.; Marine, R.; Wang, C.; Simmonds, P.; Kapusinszky, B.; Bodhidatta, L.; Oderinde, B.S.; Wommack, K.E.; Delwart, E. High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage. J. Virol. 2012, 86, 12161–12175. [Google Scholar] [CrossRef]
- Brinkman, N.E.; Fout, G.S.; Keely, S.P. Retrospective Surveillance of Wastewater To Examine Seasonal Dynamics of Enterovirus Infections. mSphere 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, M.; Martin, J. Detection by Direct Next Generation Sequencing Analysis of Emerging Enterovirus D68 and C109 Strains in an Environmental Sample From Scotland. Front. Microbiol. 2018, 9, 1956. [Google Scholar] [CrossRef]
- Majumdar, M.; Sharif, S.; Klapsa, D.; Wilton, T.; Alam, M.M.; Fernandez-Garcia, M.D.; Rehman, L.; Mujtaba, G.; McAllister, G.; Harvala, H.; et al. Environmental Surveillance Reveals Complex Enterovirus Circulation Patterns in Human Populations. Open Forum Infect. Dis. 2018, 5, ofy250. [Google Scholar] [CrossRef] [PubMed]
- Bisseux, M.; Debroas, D.; Mirand, A.; Archimbaud, C.; Peigue-Lafeuille, H.; Bailly, J.L.; Henquell, C. Monitoring of enterovirus diversity in wastewater by ultra-deep sequencing: An effective complementary tool for clinical enterovirus surveillance. Water Res. 2020, 169, 115246. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.G.; Majumdar, M.; Troman, C.; O’Toole, A.; Benny, B.; Abraham, D.; Praharaj, I.; Kang, G.; Sharif, S.; Alam, M.M.; et al. Rapid and Sensitive Direct Detection and Identification of Poliovirus from Stool and Environmental Surveillance Samples by Use of Nanopore Sequencing. J. Clin. Microbiol. 2020, 58, e00920-20. [Google Scholar] [CrossRef]
- Sadeuh-Mba, S.A.; Joffret, M.L.; Mazitchi, A.; Endegue-Zanga, M.C.; Njouom, R.; Delpeyroux, F.; Gouandjika-Vasilache, I.; Bessaud, M. Genetic and phenotypic characterization of recently discovered enterovirus D type 111. PLoS Negl. Trop. Dis. 2019, 13, e0007797. [Google Scholar] [CrossRef]
- World Health Organization. Manual for the Virological Investigation of Poliomyelitis; WHO/EP/GEN; WHO: Geneva, Switzerland, 2004. [Google Scholar]
- Diop, O.M.; Kew, O.M.; de Gourville, E.M.; Pallansch, M.A. The Global Polio Laboratory Network as a Platform for the Viral Vaccine-Preventable and Emerging Diseases Laboratory Networks. J. Infect. Dis. 2017, 216, S299–S307. [Google Scholar] [CrossRef]
- Kroiss, S.J.; Ahmadzai, M.; Ahmed, J.; Alam, M.M.; Chabot-Couture, G.; Famulare, M.; Mahamud, A.; McCarthy, K.A.; Mercer, L.D.; Muhammad, S.; et al. Assessing the sensitivity of the polio environmental surveillance system. PLoS ONE 2018, 13, e0208336. [Google Scholar] [CrossRef]
- Pons-Salort, M.; Grassly, N.C. Serotype-specific immunity explains the incidence of diseases caused by human enteroviruses. Science 2018, 361, 800–803. [Google Scholar] [CrossRef]
- Hamisu, A.W.; Blake, I.M.; Sume, G.; Braka, F.; Jimoh, A.; Dahiru, H.; Bonos, M.; Dankoli, R.; Mamuda Bello, A.; Yusuf, K.M.; et al. Characterizing Environmental Surveillance Sites in Nigeria and Their Sensitivity to Detect Poliovirus and Other Enteroviruses. J. Infect. Dis. 2020, XX, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, M.; Klapsa, D.; Wilton, T.; Akello, J.; Anscombe, C.; Allen, D.; Mee, E.T.; Minor, P.D.; Martin, J. Isolation of vaccine-like poliovirus strains in sewage samples from the UK. J. Infect. Dis. 2017, 217, 1222–1230. [Google Scholar] [CrossRef]
- Wheeler, D.L.; Church, D.M.; Lash, A.E.; Leipe, D.D.; Madden, T.L.; Pontius, J.U.; Schuler, G.D.; Schriml, L.M.; Tatusova, T.A.; Wagner, L.; et al. Database resources of the National Center for Biotechnology Information: 2002 update. Nucleic Acids Res. 2002, 30, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Kroneman, A.; Vennema, H.; Deforche, K.; Avoort, H.V.D.; Penaranda, S.; Oberste, M.S.; Vinje, J.; Koopmans, M. An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 2011, 51, 121–125. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Victoria, J.G.; Kapoor, A.; Dupuis, K.; Schnurr, D.P.; Delwart, E.L. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathog. 2008, 4, e1000163. [Google Scholar] [CrossRef]
- Fernandez-Garcia, M.D.; Majumdar, M.; Kebe, O.; Ndiaye, K.; Martin, J. Identification and whole-genome characterization of a recombinant Enterovirus B69 isolated from a patient with Acute Flaccid Paralysis in Niger, 2015. Sci. Rep. 2018, 8, 2181. [Google Scholar] [CrossRef] [PubMed]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef]
- Martin, D.P.; Murrell, B.; Khoosal, A.; Muhire, B. Detecting and Analyzing Genetic Recombination Using RDP4. Methods Mol. Biol. 2017, 1525, 433–460. [Google Scholar] [PubMed]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef]
- Padidam, M.; Sawyer, S.; Fauquet, C.M. Possible emergence of new geminiviruses by frequent recombination. Virology 1999, 265, 218–225. [Google Scholar] [CrossRef]
- Martin, D.P.; Posada, D.; Crandall, K.A.; Williamson, C. A modified bootscan algorithm for automated identification of recombinant sequences and recombination breakpoints. AIDS Res. Hum. Retrovir. 2005, 21, 98–102. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. Evaluation of methods for detecting recombination from DNA sequences: Computer simulations. Proc. Natl. Acad. Sci. USA 2001, 98, 13757–13762. [Google Scholar] [CrossRef]
- Smith, J.M. Analyzing the mosaic structure of genes. J. Mol. Evol. 1992, 34, 126–129. [Google Scholar] [CrossRef] [PubMed]
- Gibbs, M.J.; Armstrong, J.S.; Gibbs, A.J. Sister-scanning: A Monte Carlo procedure for assessing signals in recombinant sequences. Bioinformatics 2000, 16, 573–582. [Google Scholar] [CrossRef]
- Boni, M.F.; Posada, D.; Feldman, M.W. An exact nonparametric method for inferring mosaic structure in sequence triplets. Genetics 2007, 176, 1035–1047. [Google Scholar] [CrossRef]
- Faleye, T.O.C.; Adewumi, M.O.; Japhet, M.O.; David, O.M.; Oluyege, A.O.; Adeniji, J.A.; Famurewa, O. Non-polio enteroviruses in faeces of children diagnosed with acute flaccid paralysis in Nigeria. Virol. J. 2017, 14, 175. [Google Scholar] [CrossRef] [PubMed]
- Martin, J. Single Nucleotide Polymorphism Analysis of NGS Contig from EV-D111 KAT-17-1263 Strain; NIBSC: Potters Bar, UK, 2020. [Google Scholar]
- Martin, J. Single Nucleotide Polymorphism Analysis of NGS Contig from EV-D94 14-534 Strain; NIBSC: Potters Bar, UK, 2020. [Google Scholar]
- Fernandez-Garcia, M.D.; Volle, R.; Joffret, M.L.; Sadeuh-Mba, S.A.; Gouandjika-Vasilache, I.; Kebe, O.; Wiley, M.R.; Majumdar, M.; Simon-Loriere, E.; Sakuntabhai, A.; et al. Genetic Characterization of Enterovirus A71 Circulating in Africa. Emerg. Infect. Dis. 2018, 24, 754–757. [Google Scholar] [CrossRef]
- Sadeuh-Mba, S.A.; Bessaud, M.; Massenet, D.; Joffret, M.L.; Endegue, M.C.; Njouom, R.; Reynes, J.M.; Rousset, D.; Delpeyroux, F. High frequency and diversity of species C enteroviruses in Cameroon and neighboring countries. J. Clin. Microbiol. 2013, 51, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Bessaud, M.; Joffret, M.L.; Blondel, B.; Delpeyroux, F. Exchanges of genomic domains between poliovirus and other cocirculating species C enteroviruses reveal a high degree of plasticity. Sci. Rep. 2016, 6, 38831. [Google Scholar] [CrossRef]
- Burns, C.C.; Diop, O.M.; Sutter, R.W.; Kew, O.M. Vaccine-derived polioviruses. J. Infect. Dis. 2014, 210, S283–S293. [Google Scholar] [CrossRef]
- Fieldhouse, J.K.; Wang, X.; Mallinson, K.A.; Tsao, R.W.; Gray, G.C. A systematic review of evidence that enteroviruses may be zoonotic. Emerg Microbes Infect. 2018, 7, 164. [Google Scholar] [CrossRef]
- Harvala, H.; Broberg, E.; Benschop, K.; Berginc, N.; Ladhani, S.; Susi, P.; Christiansen, C.; McKenna, J.; Allen, D.; Makiello, P.; et al. Recommendations for enterovirus diagnostics and characterisation within and beyond Europe. J. Clin. Virol. 2018, 101, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Adeniji, J.A.; Faleye, T.O. Isolation and identification of enteroviruses from sewage and sewage-contaminated water in Lagos, Nigeria. Food Environ. Virol. 2014, 6, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Adeniji, J.A.; Adewale, A.O.; Faleye, T.O.C.; Adewumi, M.A. Isolation and Identification of Enteroviruses from Sewage and Sewage-Contaminated Water Samples from Ibadan, Nigeria, 2012–2013. J. Virol. Antivir. Res. 2017, 6, 75–86. [Google Scholar]
- Nikolaidis, M.; Mimouli, K.; Kyriakopoulou, Z.; Tsimpidis, M.; Tsakogiannis, D.; Markoulatos, P.; Amoutzias, G.D. Large-scale genomic analysis reveals recurrent patterns of intertypic recombination in human enteroviruses. Virology 2019, 526, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Lukashev, A.N. Role of recombination in evolution of enteroviruses. Rev. Med. Virol. 2005, 15, 157–167. [Google Scholar] [CrossRef]
- Muslin, C.; Mac Kain, A.; Bessaud, M.; Blondel, B.; Delpeyroux, F. Recombination in Enteroviruses, a Multi-Step Modular Evolutionary Process. Viruses 2019, 11, 859. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample No. | Location 1 | Collection Date |
|---|---|---|
| ADA-17-952 | Adamawa | August 2017 |
| ADA-18-059 | Adamawa | January 2018 |
| KAT-17-1263 | Katsina | October 2017 |
| NIG-17-655 | Niger | June 2017 |
| NIG-18-078 | Niger | January 2018 |
| NIG-18-202 | Niger | February 2018 |
| NIG-18-278 | Niger | March 2018 |
| NIG-18-415 | Niger | April 2018 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Majumdar, M.; Klapsa, D.; Wilton, T.; Bujaki, E.; Fernandez-Garcia, M.D.; Faleye, T.O.C.; Oyero, A.O.; Adewumi, M.O.; Ndiaye, K.; Adeniji, J.A.; et al. High Diversity of Human Non-Polio Enterovirus Serotypes Identified in Contaminated Water in Nigeria. Viruses 2021, 13, 249. https://doi.org/10.3390/v13020249
Majumdar M, Klapsa D, Wilton T, Bujaki E, Fernandez-Garcia MD, Faleye TOC, Oyero AO, Adewumi MO, Ndiaye K, Adeniji JA, et al. High Diversity of Human Non-Polio Enterovirus Serotypes Identified in Contaminated Water in Nigeria. Viruses. 2021; 13(2):249. https://doi.org/10.3390/v13020249
Chicago/Turabian StyleMajumdar, Manasi, Dimitra Klapsa, Thomas Wilton, Erika Bujaki, Maria Dolores Fernandez-Garcia, Temitope Oluwasegun Cephas Faleye, Adefunke Olufunmilayo Oyero, Moses Olubusuyi Adewumi, Kader Ndiaye, Johnson Adekunle Adeniji, and et al. 2021. "High Diversity of Human Non-Polio Enterovirus Serotypes Identified in Contaminated Water in Nigeria" Viruses 13, no. 2: 249. https://doi.org/10.3390/v13020249
APA StyleMajumdar, M., Klapsa, D., Wilton, T., Bujaki, E., Fernandez-Garcia, M. D., Faleye, T. O. C., Oyero, A. O., Adewumi, M. O., Ndiaye, K., Adeniji, J. A., & Martin, J. (2021). High Diversity of Human Non-Polio Enterovirus Serotypes Identified in Contaminated Water in Nigeria. Viruses, 13(2), 249. https://doi.org/10.3390/v13020249