
Genomic Evidence for Sequestration of Influenza A Virus Lineages in Sea Duck Host Species

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. IAV Surveillance
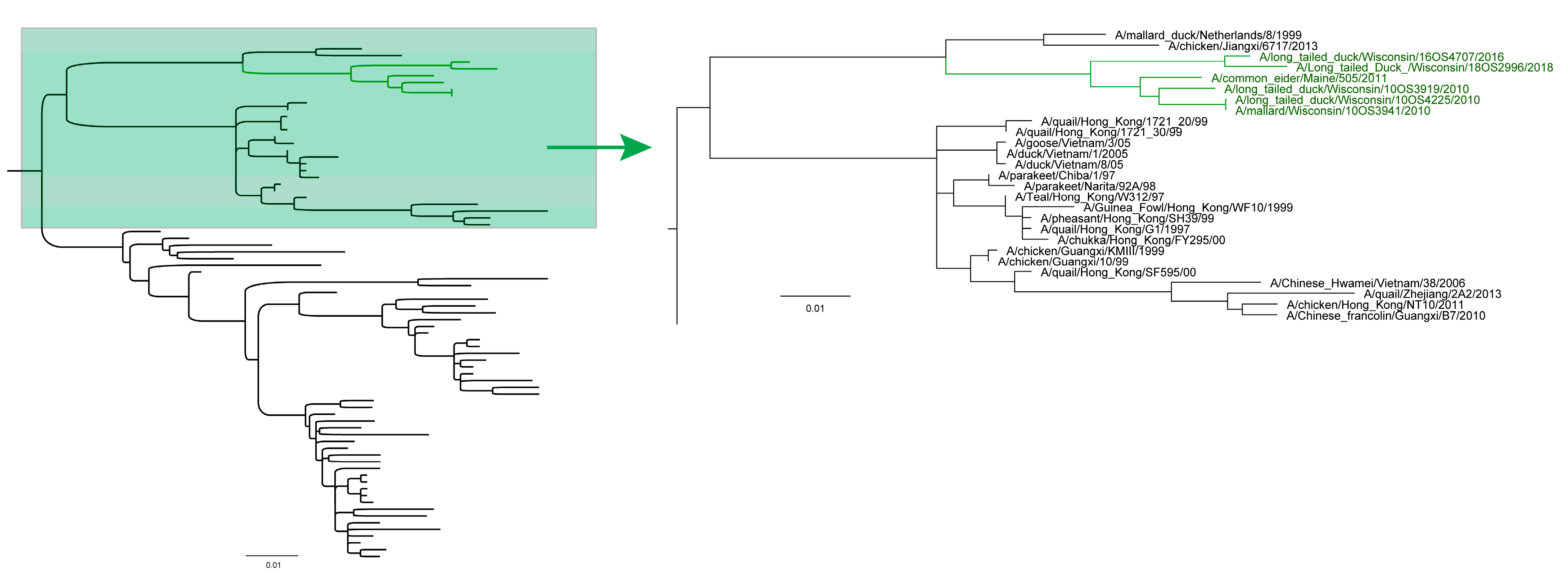
3.2. Phylogenetic Analyses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and Ecology of Influenza A Viruses. Microbiol. Mol. Biol. Rev. 1992, 56, 152–179. [Google Scholar] [CrossRef]
- Fries, A.C.; Nolting, J.M.; Danner, A.; Webster, R.G.; Bowman, A.S.; Krauss, S.; Slemons, R.D. Evidence for the Circulation and Inter-Hemispheric Movement of the H14 Subtype Influenza A Virus. PLoS ONE 2013, 8, e59216. [Google Scholar] [CrossRef] [PubMed]
- Ramey, A.M.; Pearce, J.M.; Flint, P.L.; Ip, H.S.; Derksen, D.V.; Franson, J.C.; Petrula, M.J.; Scotton, B.D.; Sowl, K.M.; Wege, M.L.; et al. Intercontinental Reassortment and Genomic Variation of Low Pathogenic Avian Influenza Viruses Isolated from Northern Pintails (Anas Acuta) in Alaska: Examining the Evidence through Space and Time. Virology 2010, 401, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Li, K.S.; Guan, Y.; Wang, J.; Smith, G.J.D.; Xu, K.M.; Duan, L.; Rahardjo, A.P.; Puthavathana, P.; Buranathai, C.; Nguyen, T.D.; et al. Genesis of a Highly Pathogenic and Potentially Pandemic H5N1 Influenza Virus in Eastern Asia. Nature 2004, 430, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Prosser, D.J.; Cui, P.; Takekawa, J.Y.; Tang, M.; Hou, Y.; Collins, B.M.; Yan, B.; Hill, N.J.; Li, T.; Li, Y.; et al. Wild Bird Migration across the Qinghai-Tibetan Plateau: A Transmission Route for Highly Pathogenic H5N1. PLoS ONE 2011, 6, e17622. [Google Scholar] [CrossRef]
- Xu, Y.; Ramey, A.M.; Bowman, A.S.; DeLiberto, T.J.; Killian, M.L.; Krauss, S.; Nolting, J.M.; Torchetti, M.K.; Reeves, A.B.; Webby, R.J.; et al. Low-Pathogenic Influenza A Viruses in North American Diving Ducks Contribute to the Emergence of a Novel Highly Pathogenic Influenza A(H7N8) Virus. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Reid, A.H.; Taubenberger, J.K. The Origin of the 1918 Pandemic Influenza Virus: A Continuing Enigma. J. Gen. Virol. 2003, 84, 2285–2292. [Google Scholar] [CrossRef]
- Kawaoka, Y.; Krauss, S.; Webster, R.G. Avian-to-Human Transmission of the PB1 Gene of Influenza A Viruses in the 1957 and 1968 Pandemics. J. Virol. 1989, 63, 4603–4608. [Google Scholar] [CrossRef]
- Smith, G.J.D.; Vijaykrishna, D.; Bahl, J.; Lycett, S.J.; Worobey, M.; Pybus, O.G.; Ma, S.K.; Cheung, C.L.; Raghwani, J.; Bhatt, S.; et al. Origins and Evolutionary Genomics of the 2009 Swine-Origin H1N1 Influenza A Epidemic. Nature 2009, 459, 1122–1125. [Google Scholar] [CrossRef]
- Webster, R.G. The Importance of Animal Influenza for Human Disease. Vaccine 2002, 20, S16–S20. [Google Scholar] [CrossRef]
- Diskin, E.R.; Friedman, K.; Krauss, S.; Nolting, J.M.; Poulson, R.L.; Slemons, R.D.; Stallknecht, D.E.; Webster, R.G.; Bowman, A.S. Subtype Diversity of Influenza A Virus in North American Waterfowl: A Multidecade Study. J. Virol. 2020, 94, e02022-19. [Google Scholar] [CrossRef] [PubMed]
- Krauss, S.; Walker, D.; Pryor, S.P.; Niles, L.; Li, C.; Hinshaw, V.S.; Webster, R.G. Influenza A Viruses of Migrating Wild Aquatic Birds in North America. Vector Borne Zoonotic Dis. 2004, 4, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Nolting, J.; Fries, A.C.; Slemons, R.D.; Courtney, C.; Hines, N.; Pedersen, J. Recovery of H14 Influenza A Virus Isolates from Sea Ducks in the Western Hemisphere. PLoS Curr. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Fries, A.C.; Nolting, J.M.; Bowman, A.S.; Killian, M.L.; Wentworth, D.E.; Slemons, R.D. Genomic Analyses Detect Eurasian-Lineage H10 and Additional H14 Influenza A Viruses Recovered from Waterfowl in the Central United States. Influenza Other Respir. Viruses 2014, 8, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Hall, J.S.; Russell, R.E.; Franson, J.C.; Soos, C.; Dusek, R.J.; Allen, R.B.; Nashold, S.W.; TeSlaa, J.L.; Jónsson, J.E.; Ballard, J.R.; et al. Avian Influenza Ecology in North Atlantic Sea Ducks: Not All Ducks Are Created Equal. PLoS ONE 2015, 10, e0144524. [Google Scholar] [CrossRef]
- Livezey, B.C. Phylogeny and Evolutionary Ecology of Modern Seaducks (Anatidae: Mergini). Condor 1995, 97, 233–255. [Google Scholar] [CrossRef]
- Sea Duck Joint Venture. Atlantic and Great Lakes Sea Duck Migration Study: Progress Report June 2015; Sea Duck Joint Venture: Anchorage, AK, USA, 2015. [Google Scholar]
- Lamb, J.S.; Paton, P.W.C.; Osenkowski, J.E.; Badzinski, S.S.; Berlin, A.M.; Bowman, T.; Dwyer, C.; Fara, L.J.; Gilliland, S.G.; Kenow, K.; et al. Spatially Explicit Network Analysis Reveals Multi-Species Annual Cycle Movement Patterns of Sea Ducks. Ecol. Appl. 2019, 29, e01919. [Google Scholar] [CrossRef]
- SDJV Sea Duck Joint Venture. Available online: https://seaduckjv.org/ (accessed on 6 December 2020).
- Fries, A.C.; Nolting, J.M.; Bowman, A.S.; Lin, X.; Halpin, R.A.; Wester, E.; Fedorova, N.; Stockwell, T.B.; Das, S.R.; Dugan, V.G.; et al. Spread and Persistence of Influenza A Viruses in Waterfowl Hosts in the North American Mississippi Migratory Flyway. J. Virol. 2015, 89, 5371–5381. [Google Scholar] [CrossRef][Green Version]
- Abente, E.J.; Gauger, P.C.; Walia, R.R.; Rajao, D.S.; Zhang, J.; Harmon, K.M.; Killian, M.L.; Vincent, A.L. Detection and Characterization of an H4N6 Avian-Lineage Influenza A Virus in Pigs in the Midwestern United States. Virology 2017, 511, 56–65. [Google Scholar] [CrossRef]
- Zhang, Y.; Aevermann, B.D.; Anderson, T.K.; Burke, D.F.; Dauphin, G.; Gu, Z.; He, S.; Kumar, S.; Larsen, C.N.; Lee, A.J.; et al. Influenza Research Database: An Integrated Bioinformatics Resource for Influenza Virus Research. Nucleic Acids Res. 2017, 45, D466–D474. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2—Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. JModelTest 2: More Models, New Heuristics and Parallel Computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Baele, G.; Lemey, P.; Bedford, T.; Rambaut, A.; Suchard, M.A.; Alekseyenko, A.V. Improving the Accuracy of Demographic and Molecular Clock Model Comparison While Accommodating Phylogenetic Uncertainty. Mol. Biol. Evol. 2012, 29, 2157–2167. [Google Scholar] [CrossRef]
- Baele, G.; Li, W.L.S.; Drummond, A.J.; Suchard, M.A.; Lemey, P. Accurate Model Selection of Relaxed Molecular Clocks in Bayesian Phylogenetics. Mol. Biol. Evol. 2013, 30, 239–243. [Google Scholar] [CrossRef]
- Drummond, A.J.; Ho, S.Y.W.; Phillips, M.J.; Rambaut, A. Relaxed Phylogenetics and Dating with Confidence. PLoS Biol. 2006, 4, e88. [Google Scholar] [CrossRef]
- Lauterbach, S.E.; McBride, D.S.; Shirkey, B.T.; Nolting, J.M.; Bowman, A.S. Year-Round Influenza a Virus Surveillance in Mallards (Anas Platyrhynchos) Reveals Genetic Persistence During the Under-Sampled Spring Season. Viruses 2020, 12, 632. [Google Scholar] [CrossRef]
- Pearce, J.M.; Reeves, A.B.; Ramey, A.M.; Hupp, J.W.; Ip, H.S.; Bertram, M.; Petrula, M.J.; Scotton, B.D.; Trust, K.A.; Meixell, B.W.; et al. Interspecific Exchange of Avian Influenza Virus Genes in Alaska: The Influence of Trans-Hemispheric Migratory Tendency and Breeding Ground Sympatry. Mol. Ecol. 2011, 20, 1015–1025. [Google Scholar] [CrossRef]
- Stallknecht, D.E.; Kearney, M.T.; Shane, S.M.; Zwank, P.J. Effects of PH, Temperature, and Salinity on Persistence of Avian Influenza Viruses in Water. Avian Dis. 1990, 34, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Breban, R.; Drake, J.M.; Stallknecht, D.E.; Rohani, P. The Role of Environmental Transmission in Recurrent Avian Influenza Epidemics. PLoS Comput. Biol. 2009, 5, e1000346. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.D.; Goekjian, G.; Poulson, R.; Valeika, S.; Stallknecht, D.E. Avian Influenza Virus in Water: Infectivity Is Dependent on PH, Salinity and Temperature. Vet. Microbiol. 2009, 136, 20–26. [Google Scholar] [CrossRef] [PubMed]
- Rohani, P.; Breban, R.; Stallknecht, D.E.; Drake, J.M. Environmental Transmission of Low Pathogenicity Avian Influenza Viruses and Its Implications for Pathogen Invasion. Proc. Natl. Acad. Sci. USA 2009, 106, 10365–10369. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Cao, B.; Hu, Y.; Feng, Z.; Wang, D.; Hu, W.; Chen, J.; Jie, Z.; Qiu, H.; Xu, K.; et al. Human Infection with a Novel Avian-Origin Influenza A (H7N9) Virus. N. Engl. J. Med. 2013, 368, 1888–1897. [Google Scholar] [CrossRef]
- Gonzalez-Reiche, A.S.; Nelson, M.I.; Angel, M.; Müller, M.L.; Ortiz, L.; Dutta, J.; van Bakel, H.; Cordon-Rosales, C.; Perez, D.R. Evidence of Intercontinental Spread and Uncommon Variants of Low-Pathogenicity Avian Influenza Viruses in Ducks Overwintering in Guatemala. mSphere 2017, 2, e00362-16. [Google Scholar] [CrossRef]
- Hurt, A.C.; Vijaykrishna, D.; Butler, J.; Baas, C.; Maurer-Stroh, S.; Silva-de-la-Fuente, M.C.; Medina-Vogel, G.; Olsen, B.; Kelso, A.; Barr, I.G.; et al. Detection of Evolutionarily Distinct Avian Influenza A Viruses in Antarctica. mBio 2014, 5, e01098-14. [Google Scholar] [CrossRef]
- Barriga, G.P.; Boric-Bargetto, D.; San Martin, M.C.; Neira, V.; van Bakel, H.; Thompsom, M.; Tapia, R.; Toro-Ascuy, D.; Moreno, L.; Vasquez, Y.; et al. Avian Influenza Virus H5 Strain with North American and Eurasian Lineage Genes in an Antarctic Penguin. Emerg. Infect. Dis. 2016, 22, 2221–2223. [Google Scholar] [CrossRef]
- Nolting, J.M.; Lauterbach, S.E.; Slemons, R.D.; Bowman, A.S. Identifying Gaps in Wild Waterfowl Influenza A Surveillance in Ohio, United States. Avian Dis. 2018, 63, 145–148. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Species | State | Total | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| DE | IL | MD | MI | MO | MS | OH | TN | WI | ||
| Black Scoter | 45(6) | 1 | 46(6) | |||||||
| Bufflehead | 6 | 21(3) | 44 | 3 | 2 | 12 | 6 | 64(8) | 158(11) | |
| Common goldeneye | 8(1) | 5 | 616(45) | 629(46) | ||||||
| Common merganser | 1 | 1 | 1 | 2 | 20 | 25 | ||||
| Hooded merganser | 5 | 10 | 5 | 2 | 2 | 14 | 14 | 1 | 20 | 73 |
| Long-tailed duck | 42 | 60 | 485(8) | 587(8) | ||||||
| Red breasted merganser | 3 | 1 | 12(1) | 16(1) | ||||||
| Surf scoter | 1 | 92(5) | 2 | 95(5) | ||||||
| White-winged scoter | 10 | 23 | 33 | |||||||
| Total | 12 | 32(3) | 239(11) | 77(1) | 2 | 16 | 34 | 7 | 1243(62) | 1662(77) |
| HA Subtype | NA Subtype | Total | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | Mixed (4,6) | Mixed (3,8) | Mixed (7,8) | ||
| 1 | 1 | 1 | 2 | ||||||||||
| 2 | 1 | 2 | 3 | ||||||||||
| 3 | 2 | 1 | 1 | 4 | |||||||||
| 4 | 2 | 2 | 30 | 34 | |||||||||
| 6 | 2 | 2 | |||||||||||
| 7 | 2 | 3 | 2 | 1 | 8 | ||||||||
| 10 | 1 | 2 | 2 | 5 | 1 | 11 | |||||||
| 11 | 6 | 6 | |||||||||||
| 12 | 2 | 2 | |||||||||||
| Mixed (4,11) | 1 | 1 | |||||||||||
| Mixed (4,10) | 1 | 1 | |||||||||||
| Total | 9 | 2 | 8 | 2 | 2 | 6 | 5 | 31 | 6 | 1 | 1 | 1 | 74 |
| Strain | PB2 | PA | HA | MP | NS |
| A/Bufflehead/Wisconsin/17OS5831/2017 | |||||
| A/Bufflehead/Wisconsin/18OS2095/2018 | |||||
| A/Bufflehead/Wisconsin/18OS3156/2018 | |||||
| A/Bufflehead/Wisconsin/18OS3297/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS3395/2018 | |||||
| A/Common goldeneye/Wisconsin/16OS3988/2016 | |||||
| A/Common goldeneye/Wisconsin/16OS4100/2016 | |||||
| A/Common goldeneye/Wisconsin/16OS4144/2016 | |||||
| A/Common goldeneye/Wisconsin/16OS4147/2016 | |||||
| A/Common goldeneye/Wisconsin/16OS4242/2016 | |||||
| A/Common goldeneye/Wisconsin/16OS4246/2016 | |||||
| A/Common goldeneye/Wisconsin/17OS5299/2017 | |||||
| A/Common goldeneye/Wisconsin/17OS5560/2017 | |||||
| A/Common goldeneye/Wisconsin/17OS5666/2017 | |||||
| A/Common goldeneye/Wisconsin/17OS5750/2017 | |||||
| A/Common goldeneye/Wisconsin/17OS5804/2017 | |||||
| A/Common goldeneye/Wisconsin/18O2145/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS2943/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS2946/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS2949/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS2961/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS2962/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS3096/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS3120/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS3144/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS3176/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS3210/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS3315/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS3317/2018 | |||||
| A/Common goldeneye/Wisconsin/18OS3389/2018 | |||||
| A/Hooded merganser/Wisconsin/17OS5556/2017 | |||||
| A/Long-tailed duck/Wisconsin/16OS4707/2016 | |||||
| A/Long-tailed duck/Wisconsin/18OS2983/2018 | |||||
| A/Long-tailed duck/Wisconsin/18OS2996/2018 | |||||
| A/Surf scoter/Maryland/15OS2532/2015 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McBride, D.S.; Lauterbach, S.E.; Li, Y.-T.; Smith, G.J.D.; Killian, M.L.; Nolting, J.M.; Su, Y.C.F.; Bowman, A.S. Genomic Evidence for Sequestration of Influenza A Virus Lineages in Sea Duck Host Species. Viruses 2021, 13, 172. https://doi.org/10.3390/v13020172
McBride DS, Lauterbach SE, Li Y-T, Smith GJD, Killian ML, Nolting JM, Su YCF, Bowman AS. Genomic Evidence for Sequestration of Influenza A Virus Lineages in Sea Duck Host Species. Viruses. 2021; 13(2):172. https://doi.org/10.3390/v13020172
Chicago/Turabian StyleMcBride, Dillon S., Sarah E. Lauterbach, Yao-Tsun Li, Gavin J. D. Smith, Mary Lea Killian, Jacqueline M. Nolting, Yvonne C. F. Su, and Andrew S. Bowman. 2021. "Genomic Evidence for Sequestration of Influenza A Virus Lineages in Sea Duck Host Species" Viruses 13, no. 2: 172. https://doi.org/10.3390/v13020172
APA StyleMcBride, D. S., Lauterbach, S. E., Li, Y.-T., Smith, G. J. D., Killian, M. L., Nolting, J. M., Su, Y. C. F., & Bowman, A. S. (2021). Genomic Evidence for Sequestration of Influenza A Virus Lineages in Sea Duck Host Species. Viruses, 13(2), 172. https://doi.org/10.3390/v13020172