
Citrus Tristeza Virus Genotype Detection Using High-Throughput Sequencing


Abstract
1. Introduction
2. Materials and Methods
2.1. Identifying Reference Sequences
2.2. Simulated Data Generation
2.3. Read Mapping
2.4. Data Visualisation
2.5. Citrus HTS Data Generation
3. Results
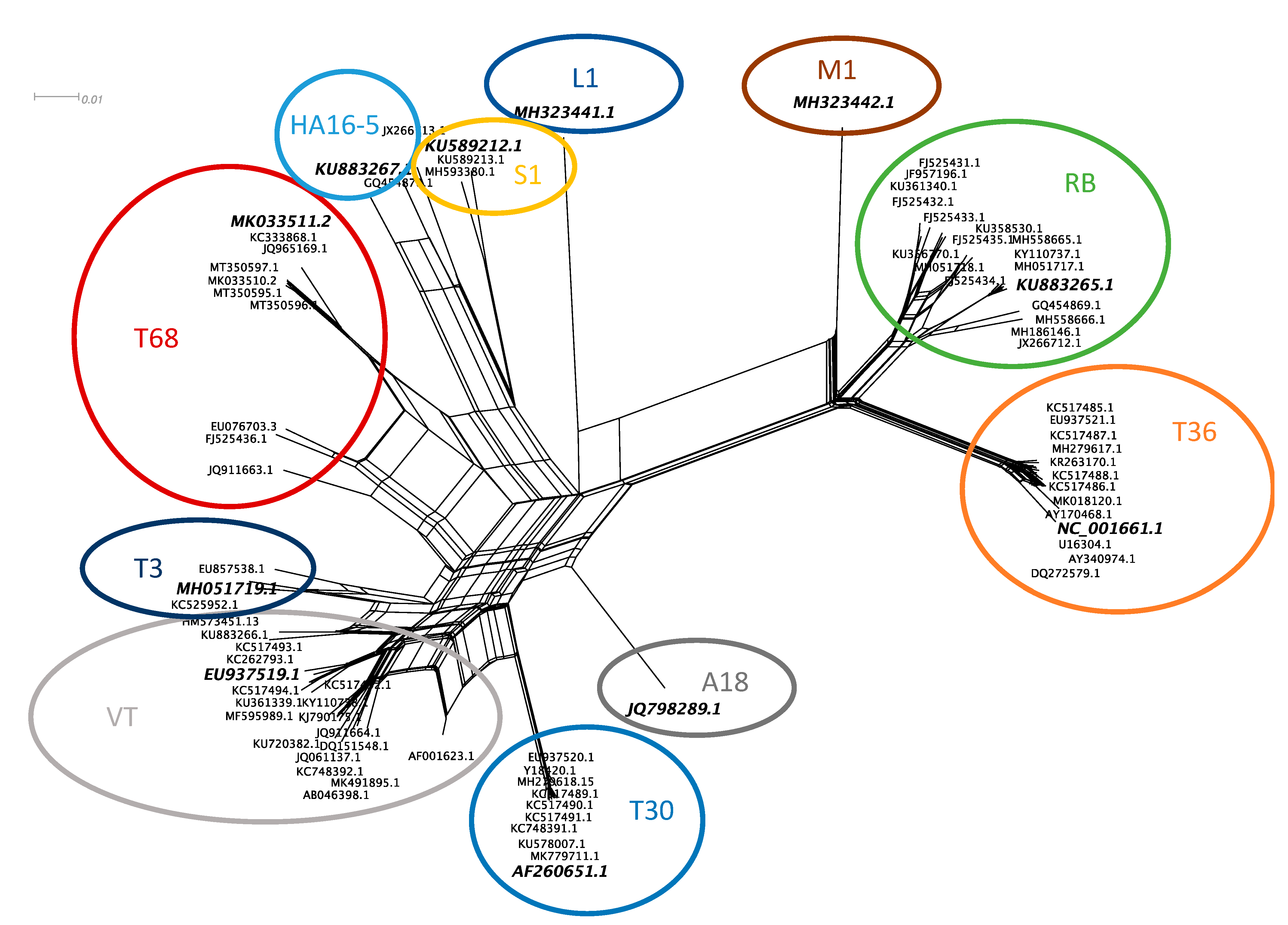
3.1. CTV Genotype Selection
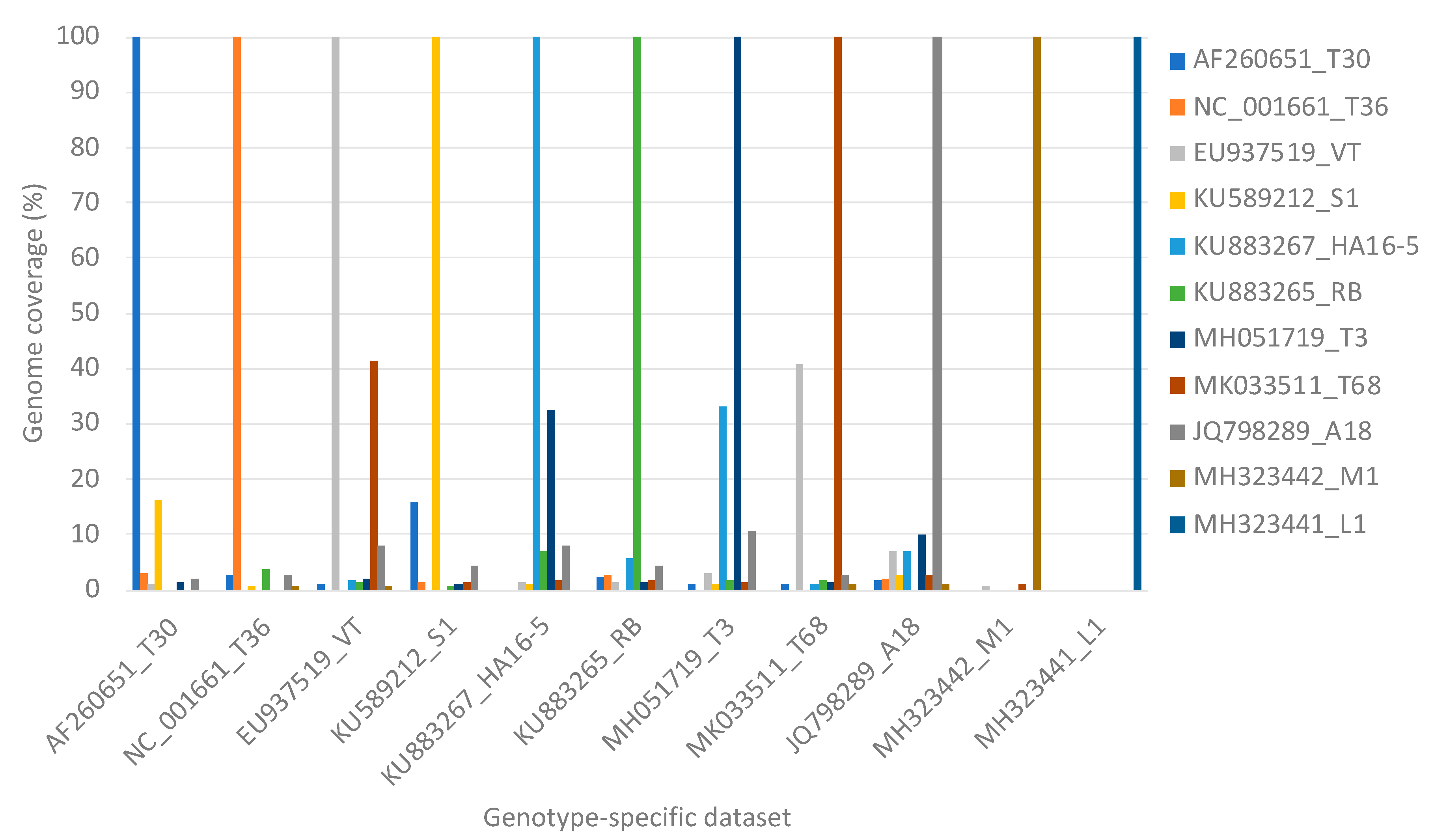
3.2. Influence of Citrus Host Reads on Virus Read Mapping
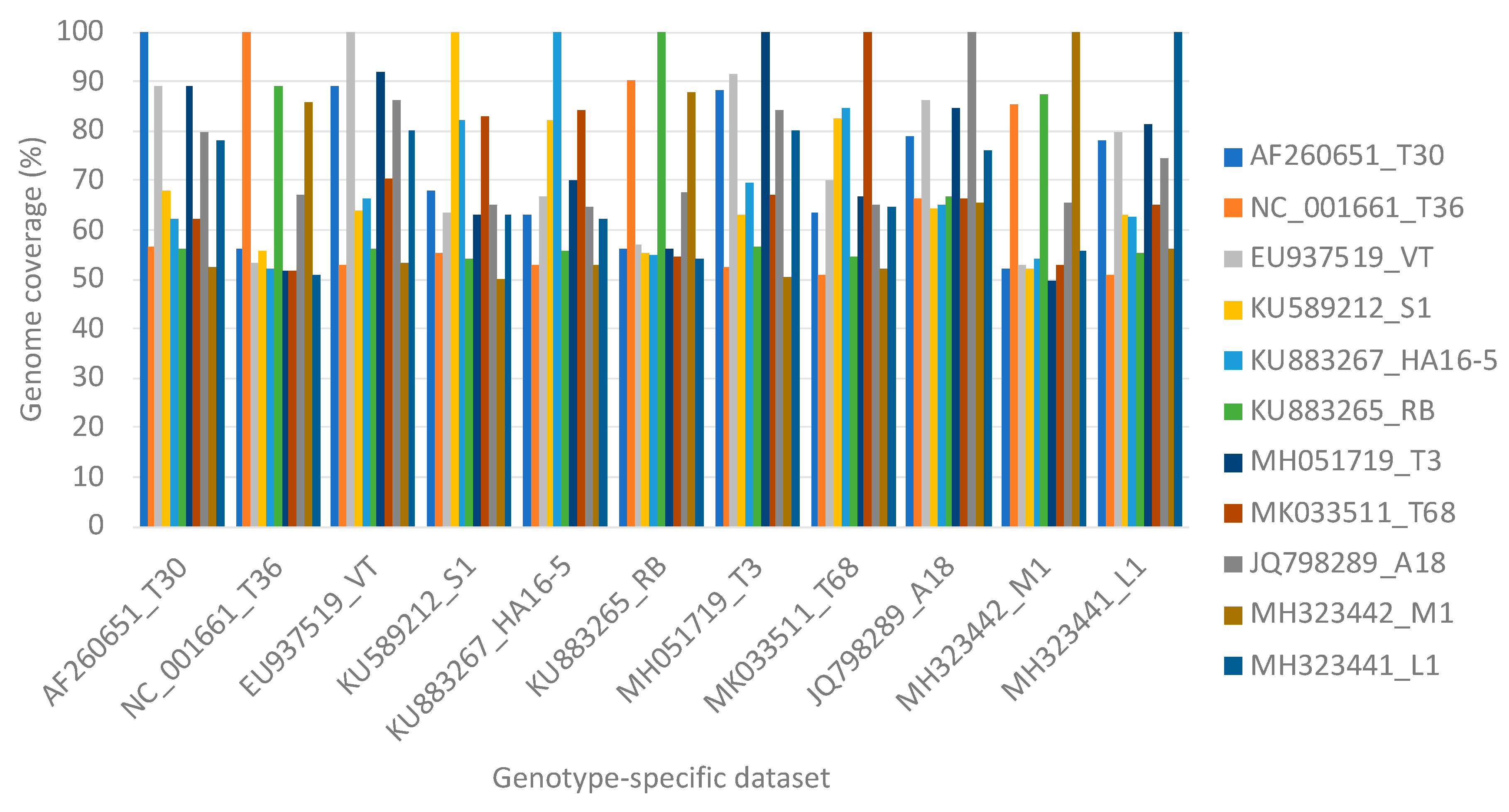
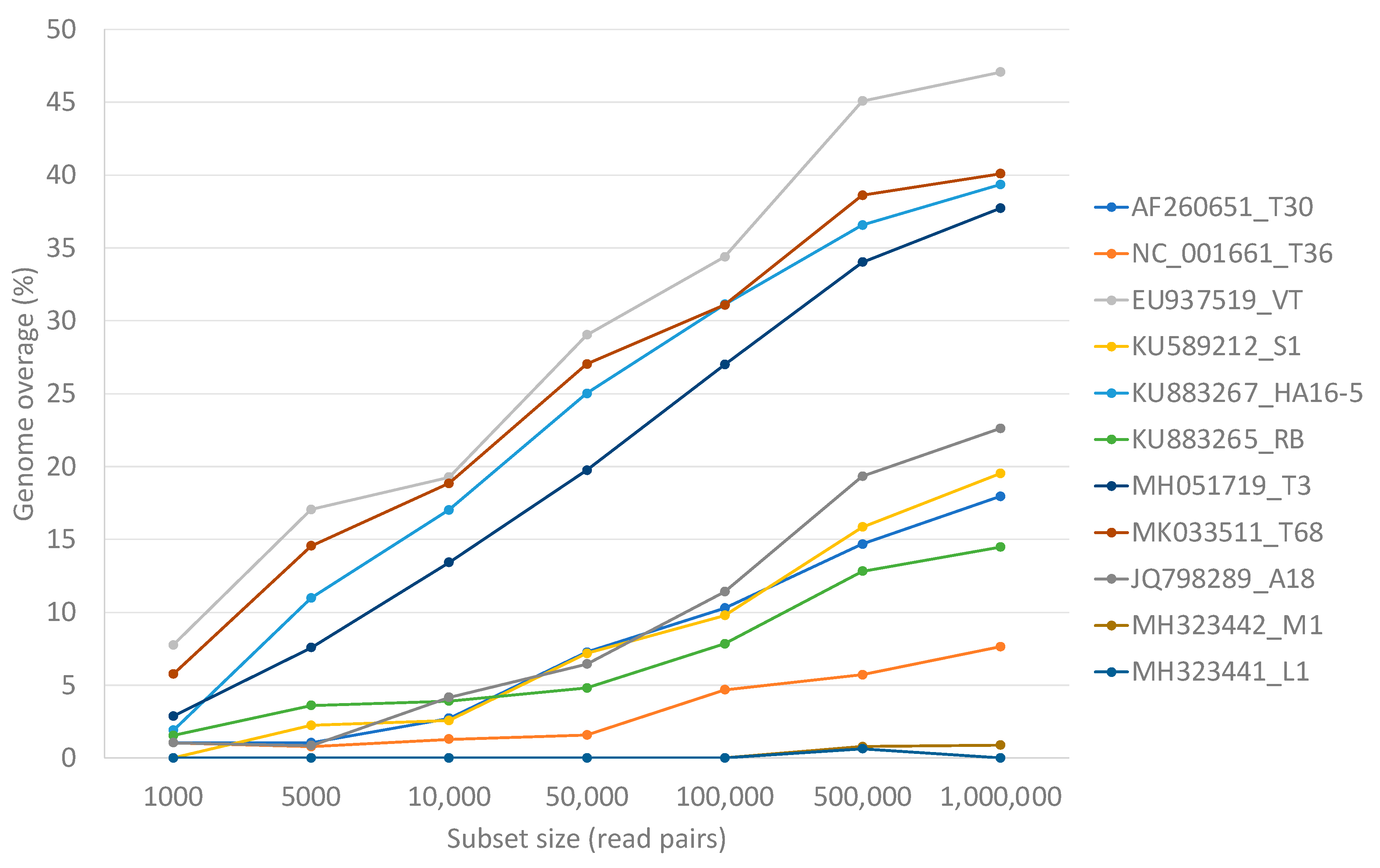
3.3. Single Genotype Infections in Variable Concentrations
3.4. Mixed Genotype Infections with Equal Concentrations of Each Genotype
3.5. Mixed Genotype Infections with Varied Concentrations
3.6. CTV Genotyping of Citrus Plants
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bombarely, A.; Rosli, H.G.; Vrebalov, J.; Moffett, P.; Mueller, L.A.; Martin, G.B. A Draft Genome Sequence of Nicotiana benthamiana to Enhance Molecular Plant-Microbe Biology Research. Mol. Plant-Microbe Interact. 2012, 25, 1523–1530. [Google Scholar] [CrossRef]
- Prochnik, S.; Marri, P.R.; Desany, B.; Rabinowicz, P.D.; Kodira, C.D.; Mohiuddin, M.; Rodriguez, F.; Fauquet, C.; Tohme, J.; Harkins, T.; et al. The Cassava Genome: Current Progress, Future Directions. Trop. Plant Biol. 2012, 5, 88–94. [Google Scholar] [CrossRef]
- van Bakel, H.; Stout, J.M.; Cote, A.G.; Tallon, C.M.; Sharpe, A.G.; Hughes, T.R.; Page, J.E. The draft genome and transcriptome of Cannabis sativa. Genome Biol. 2011, 12, R102. [Google Scholar] [CrossRef]
- Xu, Q.; Chen, L.-L.; Ruan, X.; Chen, D.; Zhu, A.; Chen, C.; Bertrand, D.; Jiao, W.-B.; Hao, B.-H.; Lyon, M.P.; et al. The draft genome of sweet orange (Citrus sinensis). Nat. Genet. 2013, 45, 59–66. [Google Scholar] [CrossRef]
- Zhang, Q.; Chen, W.; Sun, L.; Zhao, F.; Huang, B.; Yang, W.; Tao, Y.; Wang, J.; Yuan, Z.; Fan, G.; et al. The genome of Prunus mume. Nat. Commun. 2012, 3, 1318. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.P.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef]
- Enrico, E. The French–Italian Public Consortium for Grapevine Genome Characterization The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nat. Cell Biol. 2007, 449, 463–467. [Google Scholar] [CrossRef]
- Lambirth, K.; Whaley, A.M.; Blakley, I.; Schlueter, J.A.; Bost, K.L.; Loraine, A.E.; Piller, K.J. A Comparison of transgenic and wild type soybean seeds: Analysis of transcriptome profiles using RNA-Seq. BMC Biotechnol. 2015, 15, 1–17. [Google Scholar] [CrossRef]
- Cabeza, R.A.; Koester, B.; Liese, R.; Lingner, A.; Baumgarten, V.; Dirks, J.; Salinas-Riester, G.; Pommerenke, C.; Dittert, K.; Schulze, J. An RNA Sequencing Transcriptome Analysis Reveals Novel Insights into Molecular Aspects of the Nitrate Impact on the Nodule Activity of Medicago truncatula. Plant Physiol. 2013, 164, 400–411. [Google Scholar] [CrossRef]
- Zenoni, S.; Ferrarini, A.; Giacomelli, E.; Xumerle, L.; Fasoli, M.; Malerba, G.; Bellin, D.; Pezzotti, M.; Delledonne, M. Characterization of Transcriptional Complexity during Berry Development in Vitis vinifera Using RNA-Seq. Plant Physiol. 2010, 152, 1787–1795. [Google Scholar] [CrossRef]
- Cherukupalli, N.; Divate, M.; Mittapelli, S.R.; Khareedu, V.R.; Vudem, D.R. De novo Assembly of Leaf Transcriptome in the Medicinal Plant Andrographis paniculata. Front. Plant Sci. 2016, 7, 7. [Google Scholar] [CrossRef]
- Zhang, R.; Marshall, D.S.; Bryan, G.J.; Hornyik, C. Identification and Characterization of miRNA Transcriptome in Potato by High-Throughput Sequencing. PLoS ONE 2013, 8, e57233. [Google Scholar] [CrossRef]
- Bester, R.; Burger, J.T.; Maree, H.J. Transcriptome analysis reveals differentially expressed small RNAs and genes associated with grapevine leafroll-associated virus 3 infections. Physiol. Mol. Plant Pathol. 2017, 100, 220–236. [Google Scholar] [CrossRef]
- Visser, M.; Maree, H.J.; Rees, J.; Burger, J.T. High-throughput sequencing reveals small RNAs involved in ASGV infection. BMC Genom. 2014, 15, 1–10. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, P.; Li, W.; Zhang, J.; Huang, F.; Yang, J.; Bei, Y.; Lu, Y.-B. De novo transcriptome sequencing in Frankliniella occidentalis to identify genes involved in plant virus transmission and insecticide resistance. Genomics 2013, 101, 296–305. [Google Scholar] [CrossRef]
- Hofstad, A.N.; Nussbaumer, T.; Akhunov, E.; Shin, S.; Kugler, K.G.; Kistler, H.C.; Mayer, K.F.; Muehlbauer, G.J. Examining the Transcriptional Response in Wheat Fhb1 Near-Isogenic Lines to Fusarium graminearum Infection and Deoxynivalenol Treatment. Plant Genome 2016, 9. [Google Scholar] [CrossRef]
- Al-Rwahnih, M.; Daubert, S.; Golino, D.A.; Rowhani, A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef]
- Kreuze, J.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery, and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef]
- Al-Rwahnih, M.; Sudarshana, M.R.; Uyemoto, J.K.; Rowhani, A. Complete Genome Sequence of a Novel Vitivirus Isolated from Grapevine. J. Virol. 2012, 86, 9545. [Google Scholar] [CrossRef]
- Espach, Y.; Maree, H.J.; Burger, J.T. Complete Genome of a Novel Endornavirus Assembled from Next-Generation Sequence Data. J. Virol. 2012, 86, 13142. [Google Scholar] [CrossRef][Green Version]
- Villamor, D.E.V.; Mekuria, T.A.; Pillai, S.S.; Eastwell, K.C. High-Throughput Sequencing Identifies Novel Viruses in Nectarine: Insights to the Etiology of Stem-Pitting Disease. Phytopathology 2016, 106, 519–527. [Google Scholar] [CrossRef]
- Navarro, B.; Minutolo, M.; de Stradis, A.; Palmisano, F.; Alioto, D.; di Serio, F. The first phlebo-like virus infecting plants: A case study on the adaptation of negative-stranded RNA viruses to new hosts. Mol. Plant Pathology 2018, 19, 1075–1089. [Google Scholar] [CrossRef]
- Navarro, B.; Zicca, S.; Minutolo, M.; Saponari, M.; Alioto, D.; di Serio, F. A Negative-Stranded RNA Virus Infecting Citrus Trees: The Second Member of a New Genus Within the Order Bunyavirales. Front. Microbiol. 2018, 9, 2340. [Google Scholar] [CrossRef]
- Rott, M.E.; Kesanakurti, P.; Berwarth, C.; Rast, H.; Boyes, I.; Phelan, J.; Jelkmann, W. Discovery of Negative-Sense RNA Viruses in Trees Infected with Apple Rubbery Wood Disease by Next-Generation Sequencing. Plant Dis. 2018, 102, 1254–1263. [Google Scholar] [CrossRef]
- Diaz-Lara, A.; Navarro, B.; di Serio, F.; A Stevens, K.; Hwang, M.S.; Kohl, J.; Vu, S.T.; Falk, B.W.; Golino, D.A.; Al Rwahnih, M. Two Novel Negative-Sense RNA Viruses Infecting Grapevine Are Members of a Newly Proposed Genus within the Family Phenuiviridae. Viruses 2019, 11, 685. [Google Scholar] [CrossRef]
- Bester, R.; Burger, J.; Maree, H.J. Genomic characterisation of a newly identified badnavirus infecting ivy (Hedera helix). Arch. Virol. 2020, 165, 1511–1514. [Google Scholar] [CrossRef]
- Chiapello, M.; Rodríguez-Romero, J.; Nerva, L.; Forgia, M.; Chitarra, W.; Ayllón, M.A.; Turina, M. Putative new plant viruses associated with Plasmopara viticola infected grapevine samples. Ann. Appl. Biol. 2020, 176, 180–191. [Google Scholar] [CrossRef]
- Cook, G.; Coetzee, B.; Bester, R.; Breytenbach, J.H.J.; Steyn, C.; de Bruyn, R.; Burger, J.T.; Maree, H.J. Citrus Tristeza Virus Isolates of the Same Genotype Differ in Stem Pitting Severity in Grapefruit. Plant Dis. 2020, 1219–2586. [Google Scholar] [CrossRef]
- Martelli, G.P.; Abou Ghanem-Sabanadzovic, N.; Agranovsky, A.A.; Al Rwahnih, M.; Dolja, V.V.; Dovas, C.I.; Fuchs, M.; Gugerli, P.; Hu, J.S.; Jelkmann, W. Taxonomic Revision of the Family Closteroviridae with Special Reference to the Grapevine Leafroll-Associated Members of the Genus Ampelovirus and the Putative Species Unassigned to the Family. J. Plant Pathol. 2012, 94, 7–19. [Google Scholar]
- Karasev, A.; Boyko, V.; Gowda, S.; Nikolaeva, O.; Hilf, M.; Koonin, E.; Niblett, C.; Cline, K.; Gumpf, D.; Lee, R.; et al. Complete Sequence of the Citrus Tristeza Virus RNA Genome. Virology 1995, 208, 511–520. [Google Scholar] [CrossRef]
- Fuchs, M.; Bar-Joseph, M.; Candresse, T.; Maree, H.J.; Martelli, G.P.; Melzer, M.J.; Menzel, W.; Minafra, A.; Sabanadzovic, S.; Report Consortium, I. ICTV Virus Taxonomy Profile: Closteroviridae. J. Gen. Vir. 2020. [Google Scholar] [CrossRef]
- Harper, S. Citrus tristeza virus: Evolution of complex and varied genotypic groups. Front. Microbiol. 2013, 4, 93. [Google Scholar] [CrossRef]
- Cook, G.; van Vuuren, S.P.; Breytenbach, J.H.J.; Burger, J.T.; Maree, H.J. Expanded Strain-Specific RT-PCR Assay for Differential Detection of Currently Known Citrus Tristeza Virus Strains: A Useful Screening Tool. J. Phytopathol. 2016, 164, 847–851. [Google Scholar] [CrossRef]
- Yokomi, R.; Selvarajan, R.; Maheshwari, Y.; Chiumenti, M.; Saponari, M.; Giampetruzzi, A.; Weng, Z.; Xiong, Z.; Hajeri, S. Molecular and biological characterization of a novel mild strain of citrus tristeza virus in California. Arch. Virol. 2018, 163, 1795–1804. [Google Scholar] [CrossRef]
- Cook, G.; van Vuuren, S.P.; Breytenbach, J.H.J.; Steyn, C.; Burger, J.T.; Maree, H.J. Characterization of Citrus tristeza virus Single-Variant Sources in Grapefruit in Greenhouse and Field Trials. Plant Dis. 2016, 100, 2251–2256. [Google Scholar] [CrossRef]
- Moreno, P.; Ambrós, S.; Albiach-Martí, M.R.; Guerri, J.; Peña, L. Citrus tristeza virus: A pathogen that changed the course of the citrus industry. Mol. Plant Pathol. 2008, 9, 251–268. [Google Scholar] [CrossRef]
- Tatineni, S.; Robertson, C.J.; Garnsey, S.M.; Bar-Joseph, M.; Gowda, S.; Dawson, W.O. Three genes of Citrus tristeza virus are dispensable for infection and movement throughout some varieties of citrus trees. Virology 2008, 376, 297–307. [Google Scholar] [CrossRef]
- Tatineni, S.; Dawson, W.O. Enhancement or Attenuation of Disease by Deletion of Genes from Citrus Tristeza Virus. J. Virol. 2012, 86, 7850–7857. [Google Scholar] [CrossRef]
- Tatineni, S.; Robertson, C.J.; Garnsey, S.M.; Dawson, W.O. A plant virus evolved by acquiring multiple nonconserved genes to extend its host range. Proc. Natl. Acad. Sci. USA 2011, 108, 17366–17371. [Google Scholar] [CrossRef]
- Shilts, T.; el Mohtar, C.A.; Dawson, W.O.; Killiny, N. Citrus tristeza virus P33 Protein is Required for Efficient Transmission by the Aphid Aphis citricidus. Viruses 2020, 12, 1131. [Google Scholar] [CrossRef]
- Broadbent, P.; Bevington, K.B.; Coote, B.G. Control of Stem Pitting of Grapefruit in Australia by Mild Strain Protection. In Proceedings of the 11th International Organization of Citrus Virologists Conference, Orlando, FL, USA, 6–10 November 1989; University of California: Riverside, CA, USA, 1991; Volume 11, pp. 64–70. [Google Scholar]
- Salibe, A.A.; Souza, A.A.; Targon, M.L.P.N.; Müller, G.W.; Coletta Filho, H.D.; Machado, M.A. Selection of a Mild Sub-Isolate of Citrus Tristeza Virus for Preimmunization of Pera Sweet Orange. In Proceedings of the 15th International Organization of Citrus Virologists Conference, Paphos, Cyprus, 11–16 November 2001; University of California: Riverside, CA, USA, 2002; Volume 15, pp. 348–351. [Google Scholar]
- Bederski, K.; Roistacher, C.N.; Müller, G.W. Cross Protection Against the Severe Citrus Tristeza Virus Stem Pitting in Peru. In Proceedings of the 16th International Organization of Citrus Virologists Conference, Monterrey, Mexico, 7–12 November 2004; University of California: Riverside, CA, USA, 2005; Volume 16, pp. 117–125. [Google Scholar]
- da Graça, J.V.; van Vuuren, S.P. Managing citrus tristeza virus losses using cross protection. In Citrus Tristeza Virus Complex and Tristeza Diseases; American Phytopathological Society: Saint Paul, MN, USA, 2010; pp. 247–260. [Google Scholar]
- Roy, A.; Ananthakrishnan, G.; Hartung, J.S.; Brlansky, R.H. Development and Application of a Multiplex Reverse-Transcription Polymerase Chain Reaction Assay for Screening a Global Collection of Citrus tristeza virus Isolates. Phytopathology 2010, 100, 1077–1088. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.-W.; Pan, S.; Wang, G.-P.; Tang, M.; Liu, Y.; Yang, F.; Hong, N. The genotypes of citrus tristeza virus isolates from China revealed by sequence analysis of multiple molecular markers. Arch. Virol. 2012, 158, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Hilf, M.E.; Mavrodieva, V.A.; Garnsey, S.M. Genetic Marker Analysis of a Global Collection of Isolates of Citrus tristeza virus: Characterization and Distribution of CTV Genotypes and Association with Symptoms. Phytopathology 2005, 95, 909–917. [Google Scholar] [CrossRef]
- Katoh, K.; Rozewicki, J.; Yamada, K.D. MAFFT online service: Multiple sequence alignment, interactive sequence choice and visualization. Briefings Bioinform. 2019, 20, 1160–1166. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 2005, 23, 254–267. [Google Scholar] [CrossRef]
- Lole, K.S.; Bollinger, R.C.; Paranjape, R.S.; Gadkari, D.; Kulkarni, S.S.; Novak, N.G.; Ingersoll, R.; Sheppard, H.W.; Ray, S.C. Full-Length Human Immunodeficiency Virus Type 1 Genomes from Subtype C-Infected Seroconverters in India, with Evidence of Intersubtype Recombination. J. Virol. 1999, 73, 152–160. [Google Scholar] [CrossRef]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2011, 28, 593–594. [Google Scholar] [CrossRef]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Project, G.; et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Ruiz-García, A.B.; Bester, R.; Olmos, A.; Maree, H.J. Bioinformatic Tools and Genome Analysis of Citrus tristeza virus. In Citrus Tristeza Virus: Methods and Protocols; Catara, A.F., Bar-Joseph, M., Licciardello, G., Eds.; Springer: New York, NY, USA, 2019; pp. 163–178. ISBN 978-1-4939-9558-5. [Google Scholar]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.; Korobeynikov, A.; Lapidus, A.; Prjibelsky, A.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling Genomes and Mini metagenomes from Highly Chimeric Reads. In Proceedings of the Lecture Notes in Computer Science, Beijing, China, 7–10 April 2013; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2013; pp. 158–170. [Google Scholar]
- Biswas, K.K.; Tarafdar, A.; Diwedi, S.; Lee, R.F. Distribution, genetic diversity, and recombination analysis of Citrus tristeza virus of India. Virus Genes 2012, 45, 139–148. [Google Scholar] [CrossRef]
- Biswas, K.K.; Tarafdar, A.; Sharma, S.K. Complete genome sequence of mandarin decline Citrus tristeza virus of the Northeastern Himalayan hill region of India: Comparative analyses determine recombinant. Arch. Virol. 2011, 157, 579–583. [Google Scholar] [CrossRef]
- Gautom, T.; Hazarika, D.J.; Goswami, G.; Barooah, M.; Kalita, M.C.; Boro, R.C. Exploring the genetic diversity and recombination analysis of Citrus tristeza virus isolates prevalent in Northeast India. Indian Phytopathol. 2020, 73, 145–153. [Google Scholar] [CrossRef]
- Vives, M.C.; Rubio, L.; Sambade, A.; Mirkov, T.E.; Moreno, P.; Guerri, J. Evidence of multiple recombination events between two RNA sequence variants within a Citrus tristeza virus isolate. Virology 2005, 331, 232–237. [Google Scholar] [CrossRef]
- Martín, S.; Sambade, A.; Rubio, L.; Vives, M.C.; Moya, P.; Guerri, J.; Elena, S.F.; Moreno, P. Contribution of recombination and selection to molecular evolution of Citrus tristeza virus. J. Gen. Virol. 2009, 90, 1527–1538. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer Name | Polarity | Primer Sequences (5′ to 3′) | Ta (°C) 1 | Amplicon Size (bp) | Reference |
|---|---|---|---|---|---|
| CTV generic | Sense | TCTGATTGAAGTGGACGGAATAAG | 62 | 157 | [33] 2 |
| Anti-sense | GCTTAGACCAACGAGAGGATA | ||||
| RB: group1 3 | Sense | AGTGGTGGAGATTACGTTG | 60 | 628 | [33] |
| Anti-sense | TACACGCGACAAATCGAG | ||||
| RB: group 2 4 | Sense | CGGAAGGGACTACGTGGT | 60 | 658 | [33] |
| Anti-sense | CGTTTGCACGGGTTCAATG | ||||
| T36 | Sense | GGTGTAAGGAAGCGTGTGTCGCATTTA | 66 | 537 | [33] |
| Anti-sense | ACCTGCACCGTCTAACAACATCATCG | ||||
| HA16-5 | Sense | CGACAAGTGCATTACGTCTCAG | 56 | 176 | [33] |
| Anti-sense | GTAAGTATCTAAAACCAGGAG | ||||
| T68 (B165) | Sense | GTTAAGAAGGATCACCATCTTGACGTTGA | 64 | 510 | [45] 5 |
| Anti-sense | AAAATGCACTGTAACAAGACCCGACTC | ||||
| T3 | Sense | GTTATCACGCCTAAAGTTTGGTACCACT | 60 | 409 | [45] |
| Anti-sense | CATGACATCGAAGATAGCCGAAGC | ||||
| VT | Sense | TTTGAAAATGGTGATGATTTCGCCGTCA | 60 | 302 | [45] |
| Anti-sense | GACACCGGAACTGCYTGAACAGAGT | ||||
| T30 | Sense | TGTTGCGAAACTAGTTGACCCTACTG | 60 | 206 | [45] |
| Anti-sense | TAGTGGGCAGAGTGCCAAAAGAGAT | ||||
| S1 | Sense | CGACGAGTATATGAAGGACAAC | 52 | 715 | Citrus Research International (CRI) |
| Anti-sense | GAAAACCCGTAGCTGTCTAATGC |
| Genotype-Specific Data Set | Genome Coverage (%) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| AF260651 T30 | NC_001661 T36 | EU937519 VT | KU589212 S1 | KU883267 HA16-5 | KU883265 RB | MH051719 T3 | MK033511 T68 | JQ798289 A18 | MH323442 M1 | MH323441 L1 | |
| 7 | 7 | 0 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 47.1 |
| 0 | 7 | 0 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 47.7 |
| 7 | 0 | 0 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 47.7 |
| 7 | 7 | 0 | 0 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 47.7 |
| 7 | 7 | 0 | 7 | 0 | 7 | 7 | 7 | 7 | 7 | 7 | 47.7 |
| 7 | 7 | 0 | 7 | 7 | 0 | 7 | 7 | 7 | 7 | 7 | 47.6 |
| 7 | 7 | 0 | 7 | 7 | 7 | 0 | 7 | 7 | 7 | 7 | 47.7 |
| 7 | 7 | 0 | 7 | 7 | 7 | 7 | 0 | 7 | 7 | 7 | 12.8 |
| 7 | 7 | 0 | 7 | 7 | 7 | 7 | 7 | 0 | 7 | 7 | 40.9 |
| 7 | 7 | 0 | 7 | 7 | 7 | 7 | 7 | 7 | 0 | 7 | 47.1 |
| 7 | 7 | 0 | 7 | 7 | 7 | 7 | 7 | 7 | 7 | 0 | 47.1 |
| Genotype (Reference List 1) | Genome Coverage (%) | Genotype (Reference List 2) | Genome Coverage (%) |
|---|---|---|---|
| Sample 1 | |||
| AF260651_T30 | 70.5 | KC517489.1_T30_consensus | 93.8 |
| NC_001661_T36 | 40.9 | NC_001661_T36 | 38.2 |
| EU937519_VT | 99.9 | EU937519_VT | 98.9 |
| KU589212_S1 | 81.5 | KU589212_S1_consensus | 93.9 |
| KU883267_HA16-5 | 59.4 | KU883267_HA16-5_consensus | 63.2 |
| KU883265_RB | 100.0 | KU883265_RB | 100.0 |
| MH051719_T3 | 98.8 | MH051719_T3 | 98.4 |
| MK033511_T68 | 59.4 | MK033511_T68_consensus | 69.0 |
| JQ798289_A18 | 68.3 | JQ798289_A18_consensus | 92.3 |
| MH323442_M1 | 24.7 | MH323442_M1 | 24.7 |
| MH323441_L1 | 17.1 | MH323441_L1 | 15.3 |
| Sample 2 | |||
| AF260651_T30 | 6.5 | ||
| NC_001661_T36 | 2.8 | ||
| EU937519_VT | 43.8 | ||
| KU589212_S1 | 13.4 | ||
| KU883267_HA16-5 | 12.6 | ||
| KU883265_RB | 39.0 | ||
| MH051719_T3 | 21.6 | ||
| MK033511_T68 | 100.0 | ||
| JQ798289_A18 | 16.3 | ||
| MH323442_M1 | 2.9 | ||
| MH323441_L1 | 2.1 | ||
| Sample 3 | |||
| AF260651_T30 | 62.3 | KC517489.1_T30_consensus | 92.8 |
| NC_001661_T36 | 33.9 | NC_001661_T36 | 34.1 |
| EU937519_VT | 99.9 | EU937519_VT | 95.3 |
| KU589212_S1 | 77.1 | KU589212_S1_consensus | 90.6 |
| KU883267_HA16-5 | 55.5 | KU883267_HA16-5_consensus | 59.7 |
| KU883265_RB | 100.0 | KU883265_RB | 100.0 |
| MH051719_T3 | 98.2 | MH051719_T3 | 96.8 |
| MK033511_T68 | 60.9 | MK033511_T68_consensus | 67.5 |
| JQ798289_A18 | 58.5 | JQ798289_A18_consensus | 88.7 |
| MH323442_M1 | 19.0 | MH323442_M1 | 18.7 |
| MH323441_L1 | 11.0 | MH323441_L1 | 7.7 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bester, R.; Cook, G.; Maree, H.J. Citrus Tristeza Virus Genotype Detection Using High-Throughput Sequencing. Viruses 2021, 13, 168. https://doi.org/10.3390/v13020168
Bester R, Cook G, Maree HJ. Citrus Tristeza Virus Genotype Detection Using High-Throughput Sequencing. Viruses. 2021; 13(2):168. https://doi.org/10.3390/v13020168
Chicago/Turabian StyleBester, Rachelle, Glynnis Cook, and Hans J. Maree. 2021. "Citrus Tristeza Virus Genotype Detection Using High-Throughput Sequencing" Viruses 13, no. 2: 168. https://doi.org/10.3390/v13020168
APA StyleBester, R., Cook, G., & Maree, H. J. (2021). Citrus Tristeza Virus Genotype Detection Using High-Throughput Sequencing. Viruses, 13(2), 168. https://doi.org/10.3390/v13020168