
Induction of Brain Tumors by the Archetype Strain of Human Neurotropic JCPyV in a Transgenic Mouse Model



Abstract
1. Introduction
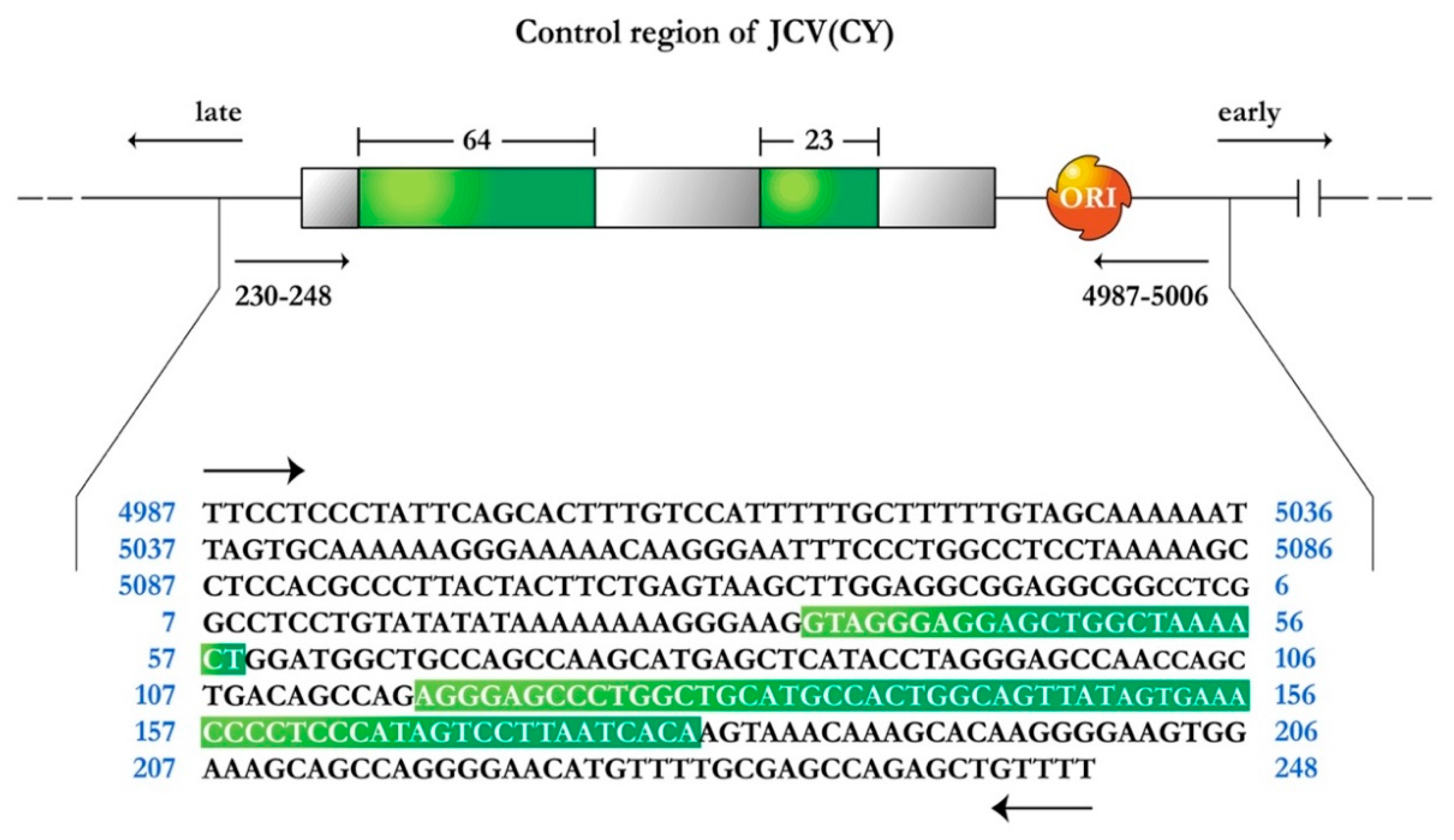
1.1. JCPyV Genomic Structure
1.2. JCPyV Oncogenicity
2. Materials and Methods
2.1. Development of Transgenic Mice
2.2. Histology and Immunohistochemistry
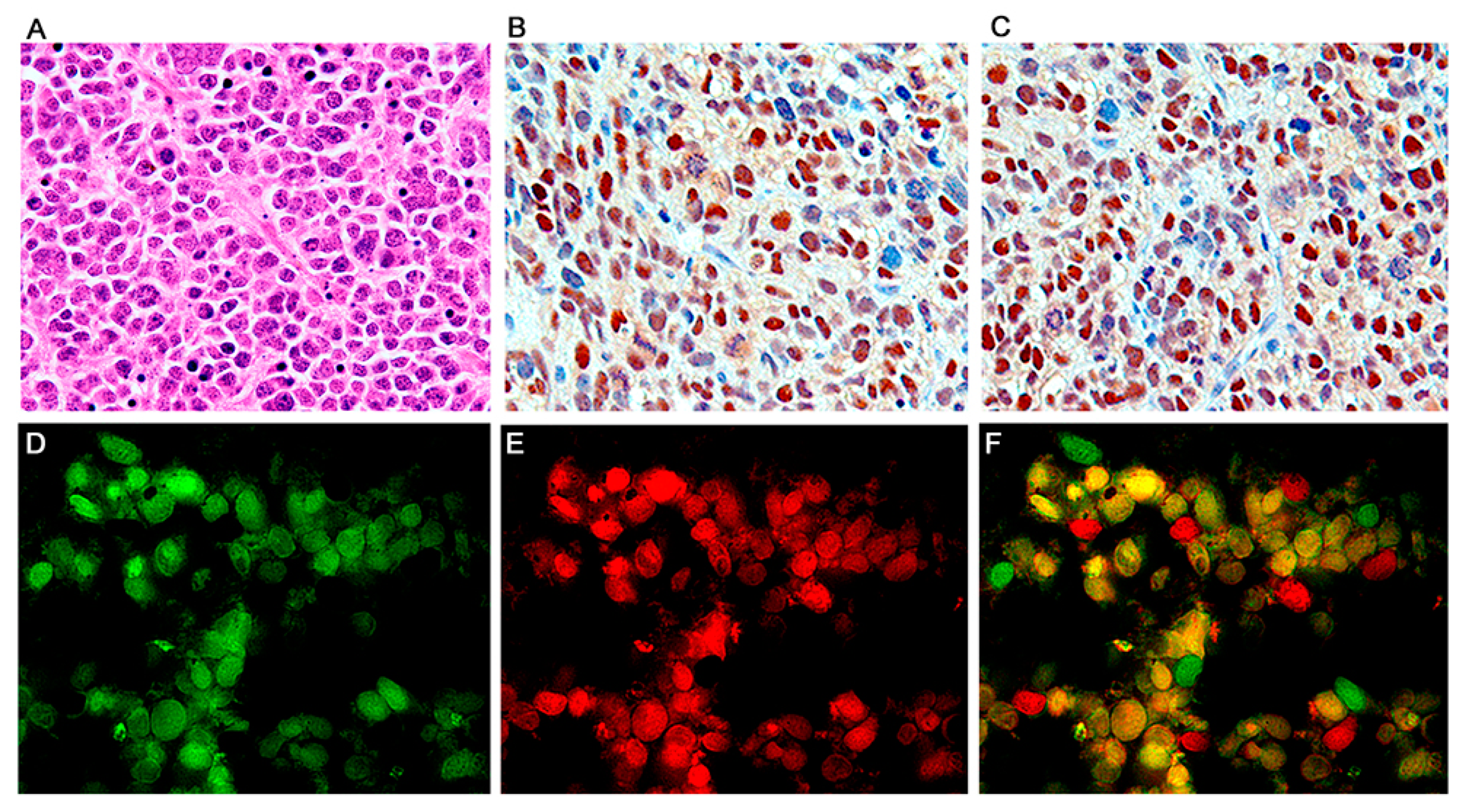
2.3. Double Labeling Immunofluorescence
2.4. Protein Extraction and Western Blot Analysis
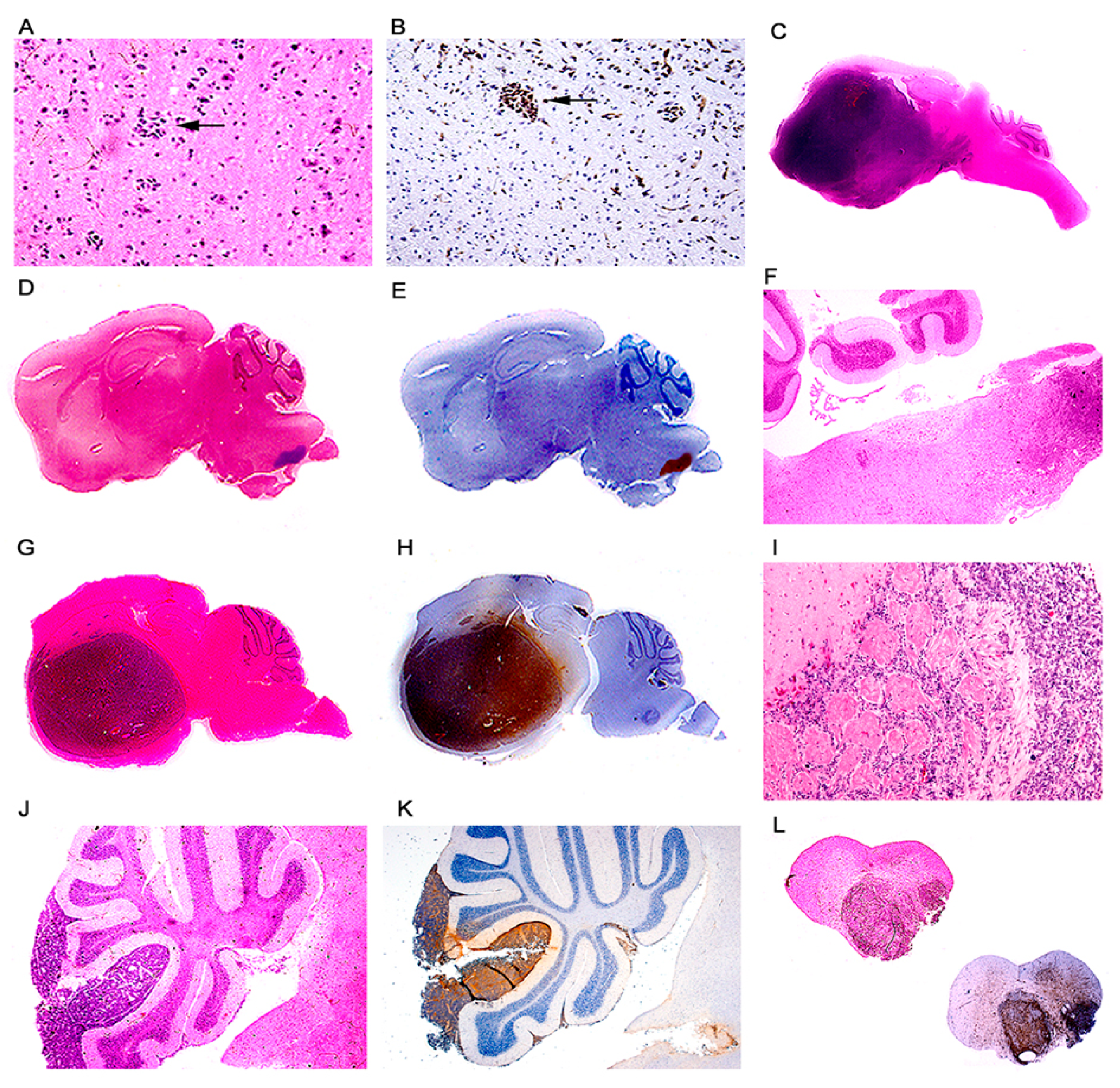
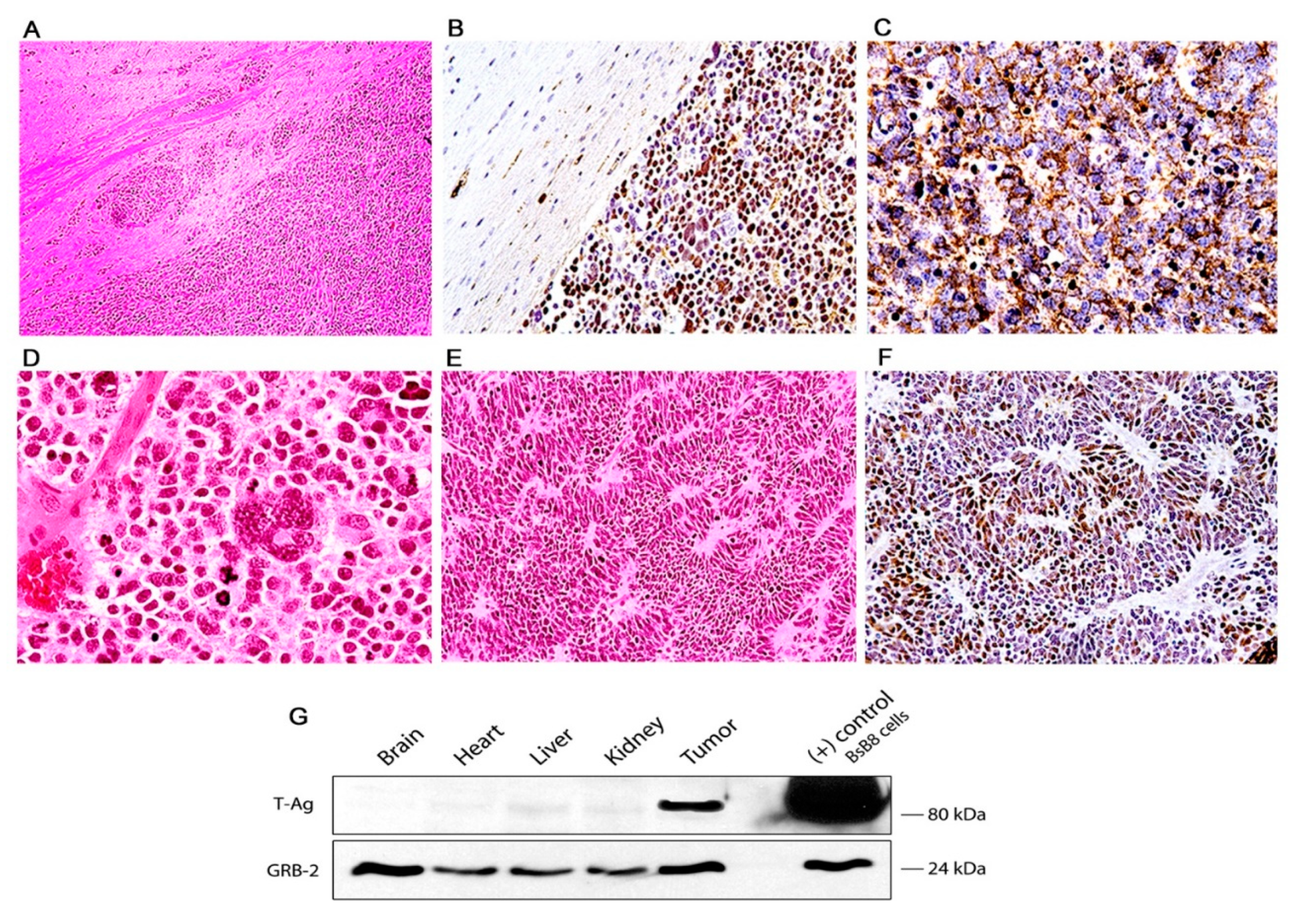
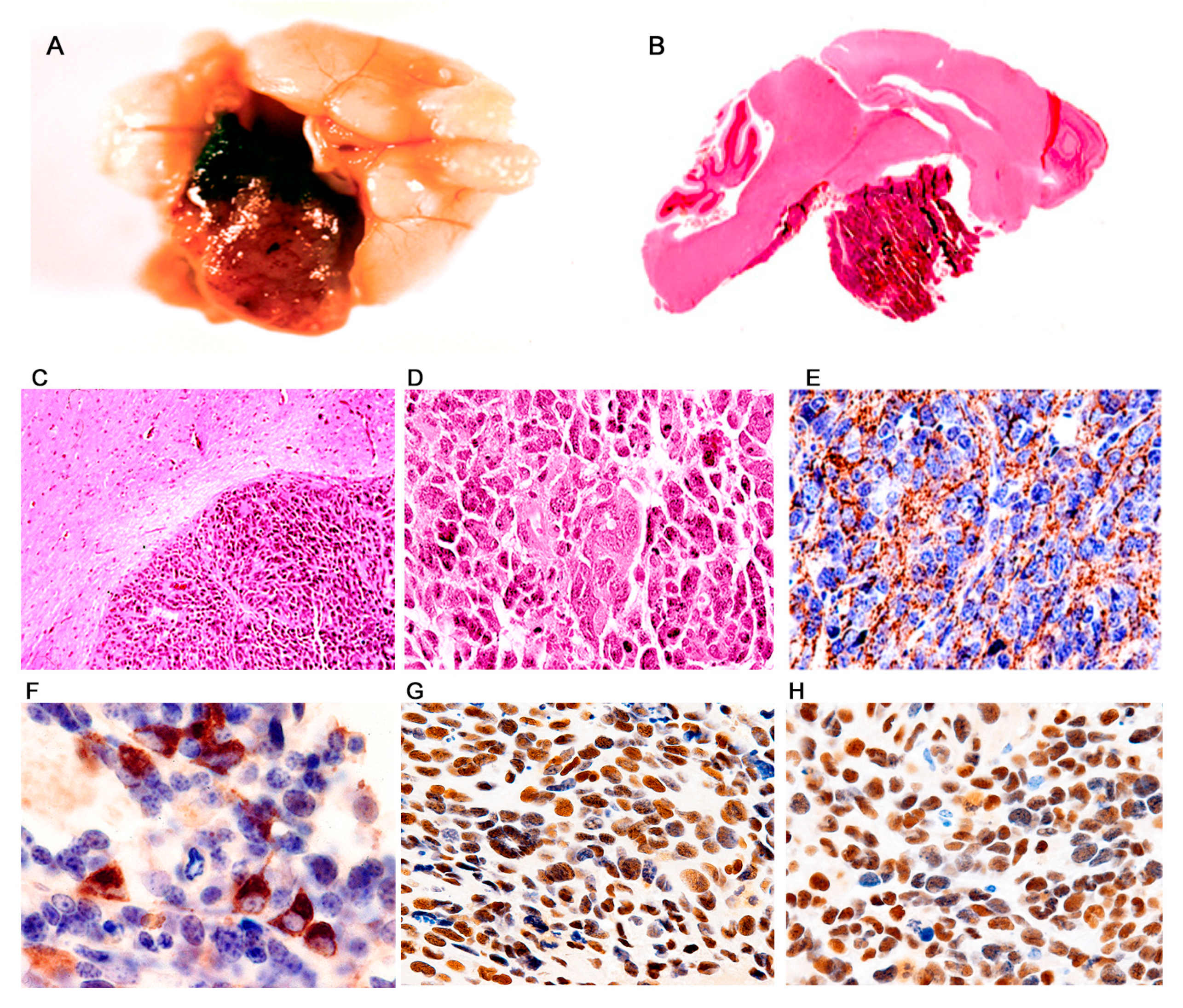
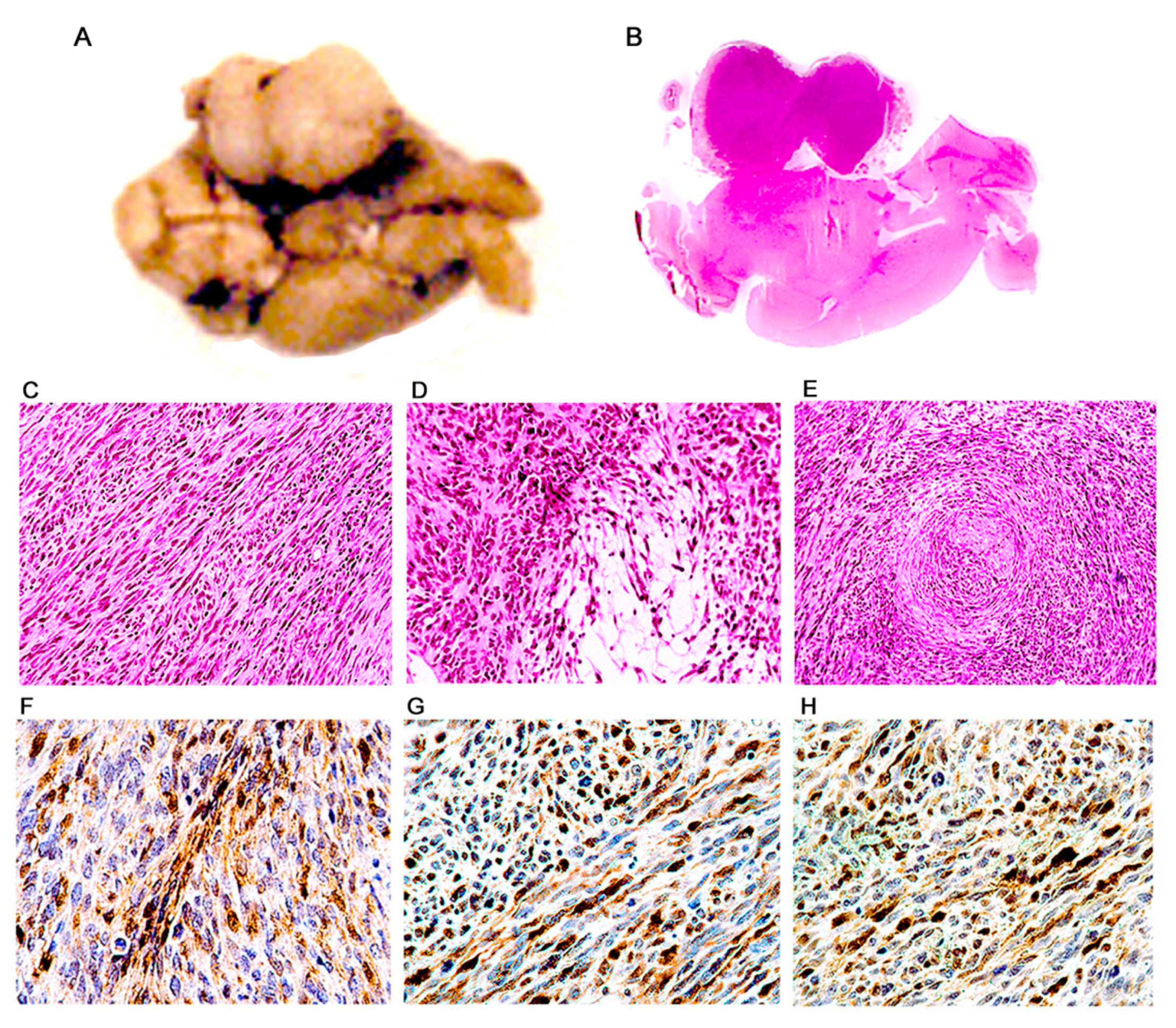
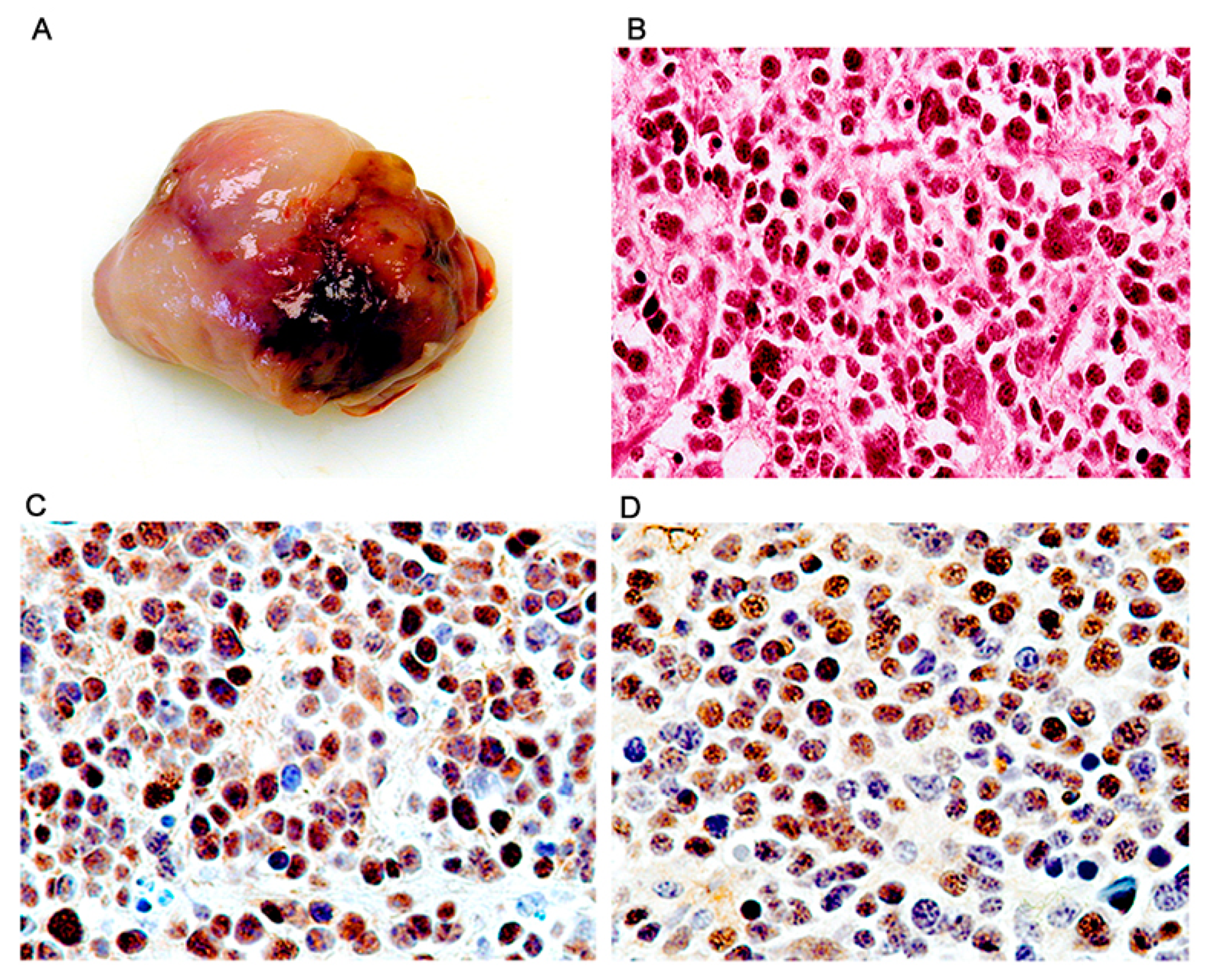
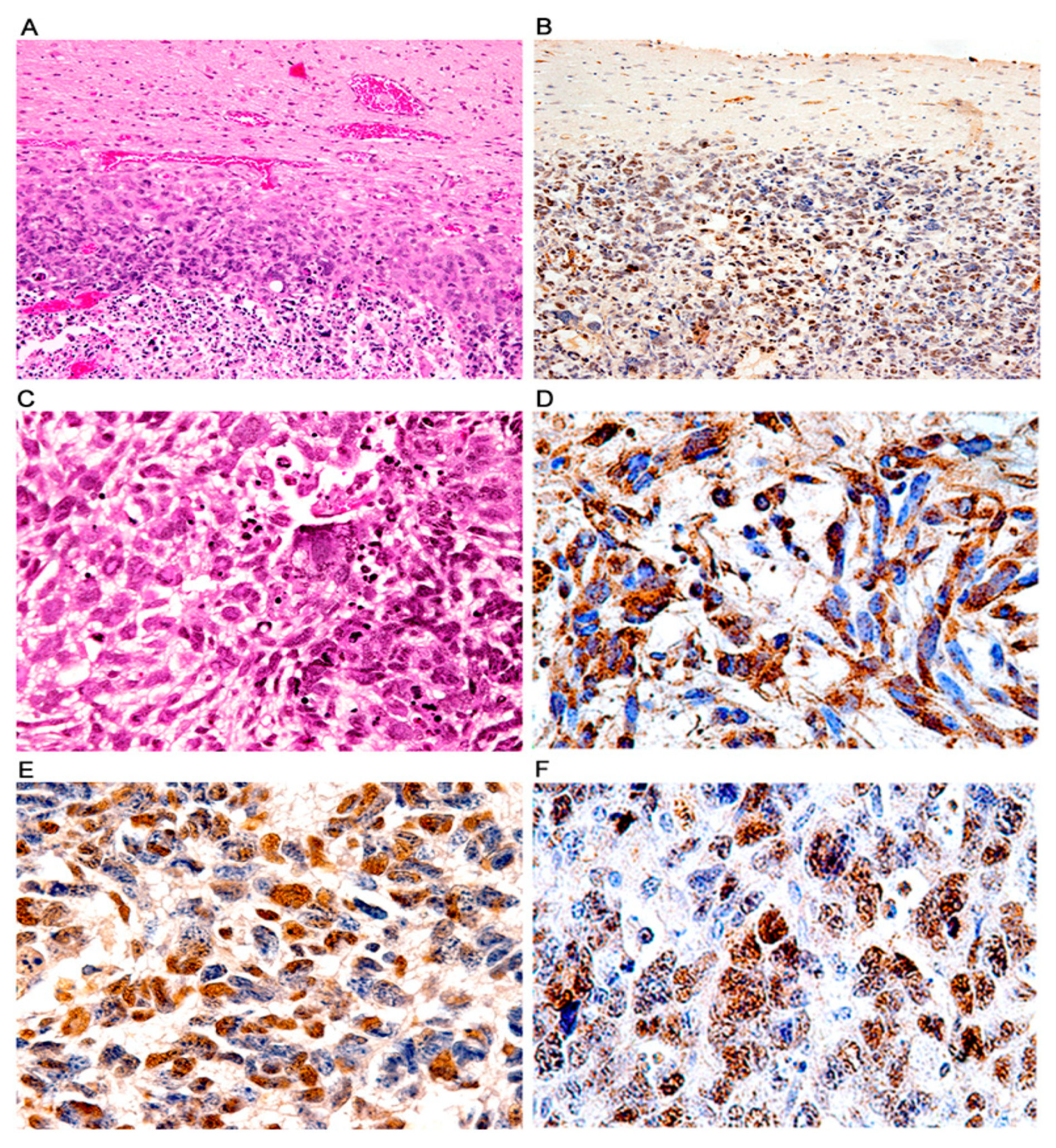
3. Results
Characterization of Tumors in CY Archetype Transgenic Mice
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Moens, U.; Calvignac-Spencer, S.; Lauber, C.; Ramqvist, T.; Feltkamp, M.C.W.; Daugherty, M.D.; Verschoor, E.J.; Ehlers, B.; Ictv Report, C. ICTV Virus taxonomy profile: Polyomaviridae. J. Gen. Virol. 2017, 98, 1159–1160. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.L.; Padgett, B.L. The epidemiology of human Polyomaviruses. Prog. Clin. Biol Res. 1983, 105, 99–106. [Google Scholar] [PubMed]
- Taguchi, F.; Kajioka, J.; Miyamura, T. Prevalence rate and age of acquisition of antibodies against JC Virus and BK Virus in human sera. Microbiol. Immunol. 1982, 26, 1057–1064. [Google Scholar] [CrossRef] [PubMed]
- Monaco, M.C.; Jensen, P.N.; Hou, J.; Durham, L.C.; Major, E.O. Detection of JC virus DNA in human tonsil tissue: Evidence for site of initial viral infection. J. Virol. 1998, 72, 9918–9923. [Google Scholar] [CrossRef] [PubMed]
- Wei, G.; Liu, C.K.; Atwood, W.J. JC virus binds to primary human glial cells, tonsillar stromal cells, and B-lymphocytes, but not to T lymphocytes. J. NeuroVirol. 2000, 6, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Bofill-Mas, S.; Pina, S.; Girones, R. Documenting the epidemiologic patterns of Polyomaviruses in human populations by studying their presence in urban sewage. Appl. Env. Microbiol. 2000, 66, 238–245. [Google Scholar] [CrossRef]
- Bofill-Mas, S.; Formiga-Cruz, M.; Clemente-Casares, P.; Calafell, F.; Girones, R. Potential transmission of human Polyomaviruses through the gastrointestinal tract after exposure to virions or viral DNA. J. Virol. 2001, 75, 10290–10299. [Google Scholar] [CrossRef]
- Ricciardiello, L.; Laghi, L.; Ramamirtham, P.; Chang, C.L.; Chang, D.K.; Randolph, A.E.; Boland, C.R. JC Virus DNA sequences are frequently present in the human upper and lower gastrointestinal tract. Gastroenterology 2000, 119, 1228–1235. [Google Scholar] [CrossRef]
- Laghi, L.; Randolph, A.E.; Chauhan, D.P.; Marra, G.; Major, E.O.; Neel, J.V.; Boland, C.R. JC Virus DNA is present in the mucosa of the human colon and in colorectal cancers. Proc. Natl. Acad. Sci. USA 1999, 96, 7484–7489. [Google Scholar] [CrossRef]
- Abreu, I.N.; Cortinhas, J.M.; Dos Santos, M.B.; Queiroz, M.A.F.; da Silva, A.; Cayres-Vallinoto, I.M.V.; Vallinoto, A.C.R. Detection of Human Polyomavirus 2 (HPyV2) in oyster samples in northern Brazil. Virol. J. 2020, 17, 85. [Google Scholar] [CrossRef]
- Coleman, D.V.; Daniel, R.A.; Gardner, S.D.; Field, A.M.; Gibson, P.E. Polyoma Virus in urine during pregnancy. Lancet 1977, 2, 709–710. [Google Scholar] [CrossRef]
- Markowitz, R.B.; Eaton, B.A.; Kubik, M.F.; Latorra, D.; McGregor, J.A.; Dynan, W.S. BK Virus and JC Virus shed during pregnancy have predominantly archetypal regulatory regions. J. Virol. 1991, 65, 4515–4519. [Google Scholar] [CrossRef] [PubMed]
- Major, E.O.; Amemiya, K.; Tornatore, C.S.; Houff, S.A.; Berger, J.R. Pathogenesis and molecular biology of Progressive Multifocal Leukoencephalopathy, the JC virus-induced demyelinating disease of the human brain. Clin. Microbiol. Rev. 1992, 5, 49–73. [Google Scholar] [CrossRef]
- Berger, J.R.; Concha, M. Progressive Multifocal Leukoencephalopathy: The evolution of a disease once considered rare. J. NeuroVirol. 1995, 1, 5–18. [Google Scholar] [CrossRef] [PubMed]
- Miller, J.R.; Barrett, R.E.; Britton, C.B.; Tapper, M.L.; Bahr, G.S.; Bruno, P.J.; Marquardt, M.D.; Hays, A.P.; McMurtry, J.G., 3rd; Weissman, J.B.; et al. Progressive Multifocal Leukoencephalopathy in a male homosexual with T-cell immune deficiency. N. Engl. J. Med. 1982, 307, 1436–1438. [Google Scholar] [CrossRef] [PubMed]
- Holman, R.C.; Torok, T.J.; Belay, E.D.; Janssen, R.S.; Schonberger, L.B. Progressive Multifocal Leukoencephalopathy in the United States, 1979–1994: Increased mortality associated with HIV infection. Neuroepidemiology 1998, 17, 303–309. [Google Scholar] [CrossRef]
- Aksamit, A.J., Jr. Progressive Multifocal Leukoencephalopathy: A review of the pathology and pathogenesis. Microsc. Res. Tech. 1995, 32, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, L.; Piña-Oviedo, S. HIV disorders of the brain: Pathology and pathogenesis. Front. Biosci. 2006, 11, 718–732. [Google Scholar] [CrossRef]
- Frisque, R.J.; Bream, G.L.; Cannella, M.T. Human Polyomavirus JC virus genome. J. Virol. 1984, 51, 458–469. [Google Scholar] [CrossRef]
- Raj, G.V.; Khalili, K. Transcriptional regulation: Lessons from the human neurotropic Polyomavirus, JCV. Virology 1995, 213, 283–291. [Google Scholar] [CrossRef]
- Yogo, Y.; Kitamura, T.; Sugimoto, C.; Ueki, T.; Aso, Y.; Hara, K.; Taguchi, F. Isolation of a possible archetypal JC virus DNA sequence from non-immunocompromised individuals. J. Virol. 1990, 64, 3139–3143. [Google Scholar] [CrossRef] [PubMed]
- Åström, K.E.; Mancall, E.L.; Richardson, E.P., Jr. Progressive Multifocal Leuko-encephalopathy; a hitherto unrecognized complication of chronic lymphatic leukaemia and Hodgkin’s disease. Brain 1958, 81, 93–111. [Google Scholar] [CrossRef] [PubMed]
- Richardson, E.P., Jr. Progressive Multifocal Leukoencephalopathy. N. Engl. J. Med. 1961, 265, 815–823. [Google Scholar] [CrossRef]
- Silverman, L.; Rubinstein, L.J. Electron microscopic observations on a case of Progressive Multifocal Leukoencephalopathy. Acta NeuroPathol. 1965, 5, 215–224. [Google Scholar] [CrossRef] [PubMed]
- Zu Rhein, G.; Chou, S.M. Particles resembling Papova viruses in human cerebral demyelinating disease. Science 1965, 148, 1477–1479. [Google Scholar] [CrossRef]
- Padgett, B.L.; Walker, D.L.; Zu Rhein, G.M.; Eckroade, R.J.; Dessel, B.H. Cultivation of Papova-like virus from human brain with Progressive Multifocal Leucoencephalopathy. Lancet 1971, 1, 1257–1260. [Google Scholar] [CrossRef]
- Walker, D.L.; Padgett, B.L.; Zu Rhein, G.M.; Albert, A.E.; Marsh, R.F. Human Papovavirus (JC): Induction of brain tumors in hamsters. Science 1973, 181, 674–676. [Google Scholar] [CrossRef]
- Zu Rhein, G.M.; Varakis, J.N. Perinatal induction of medulloblastomas in Syrian golden hamsters by a human polyoma virus (JC). Natl. Cancer Inst. Monogr. 1979, 51, 205–208. [Google Scholar]
- Varakis, J.; Zu Rhein, G.M.; Padgett, B.L.; Walker, D.L. Induction of peripheral neuroblastomas in Syrian hamsters after injection as neonates with JC virus, a human polyoma virus. Cancer Res. 1978, 38, 1718–1722. [Google Scholar]
- Ohsumi, S.; Ikehara, I.; Motoi, M.; Ogawa, K.; Nagashima, K.; Yasui, K. Induction of undifferentiated brain tumors in rats by a human Polyomavirus (JC virus). Jpn. J. Cancer Res. 1985, 76, 429–431. [Google Scholar]
- Ohsumi, S.; Motoi, M.; Ogawa, K. Induction of undifferentiated tumors by JC Virus in the cerebrum of rats. Acta Pathol. Jpn. 1986, 36, 815–825. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, K.; Yasui, K.; Kimura, J.; Washizu, M.; Yamaguchi, K.; Mori, W. Induction of brain tumors by a newly isolated JC virus (Tokyo-1 strain). Am. J. Pathol. 1984, 116, 455–463. [Google Scholar]
- London, W.T.; Houff, S.A.; Madden, D.L.; Fuccillo, D.A.; Gravell, M.; Wallen, W.C.; Palmer, A.E.; Sever, J.L.; Padgett, B.L.; Walker, D.L.; et al. Brain tumors in owl monkeys inoculated with a human Polyomavirus (JC virus). Science 1978, 201, 1246–1249. [Google Scholar] [CrossRef] [PubMed]
- London, W.T.; Houff, S.A.; McKeever, P.E.; Wallen, W.C.; Sever, J.L.; Padgett, B.L.; Walker, D.L. Viral-induced astrocytomas in squirrel monkeys. Prog. Clin. Biol Res. 1983, 105, 227–237. [Google Scholar]
- O’Neill, F.J.; Frisque, R.J.; Xu, X.; Hu, Y.X.; Carney, H. Immortalization of human cells by mutant and chimeric primate polyomavirus T-Antigen genes. Oncogene 1995, 10, 1131–1139. [Google Scholar] [PubMed]
- Khalili, K.; Krynska, B.; Del Valle, L.; Katsetos, C.D.; Croul, S. Medulloblastomas and the human neurotropic polyomavirus JC virus. Lancet 1999, 353, 1152–1153. [Google Scholar] [CrossRef]
- Krynska, B.; Del Valle, L.; Croul, S.; Gordon, J.; Katsetos, C.D.; Carbone, M.; Giordano, A.; Khalili, K. Detection of human neurotropic JC virus DNA sequence and expression of the viral oncogenic protein in pediatric medulloblastomas. Proc. Natl. Acad Sci. USA 1999, 96, 11519–11524. [Google Scholar] [CrossRef] [PubMed]
- Del Valle, L.; Gordon, J.; Enam, S.; Delbue, S.; Croul, S.; Abraham, S.; Radhakrishnan, S.; Assimakopoulou, M.; Katsetos, C.D.; Khalili, K. Expression of human neurotropic Polyomavirus JCV late gene product Agnoprotein in human Medulloblastoma. J. Natl. Cancer Inst. 2002, 94, 267–273. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Caldarelli-Stefano, R.; Boldorini, R.; Monga, G.; Meraviglia, E.; Zorini, E.O.; Ferrante, P. JC Virus in human glial-derived tumors. Hum. Pathol. 2000, 31, 394–395. [Google Scholar] [CrossRef]
- Del Valle, L.; Gordon, J.; Assimakopoulou, M.; Enam, S.; Geddes, J.F.; Varakis, J.N.; Katsetos, C.D.; Croul, S.; Khalili, K. Detection of JC Virus DNA sequences and expression of the viral regulatory protein T-Antigen in tumors of the Central Nervous System. Cancer Res. 2001, 61, 4287–4293. [Google Scholar]
- Del Valle, L.; Azizi, S.A.; Krynska, B.; Enam, S.; Croul, S.E.; Khalili, K. Reactivation of human neurotropic JC Virus expressing oncogenic protein in a recurrent Glioblastoma multiforme. Ann. Neurol. 2000, 48, 932–936. [Google Scholar] [CrossRef]
- Piña-Oviedo, S.; De Leon-Bojorge, B.; Cuesta-Mejias, T.; White, M.K.; Ortiz-Hidalgo, C.; Khalili, K.; Del Valle, L. Glioblastoma multiforme with small cell neuronal-like component: Association with human neurotropic JC Virus. Acta NeuroPathol. 2006, 111, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Mineta, T.; Ueda, S.; Nakahara, Y.; Shiraishi, T.; Tamiya, T.; Tabuchi, K. Detection of JC Virus DNA sequences in brain tumors in pediatric patients. J. Neurosurg. 2005, 102 (Suppl. 3), 294–298. [Google Scholar] [CrossRef]
- Del Valle, L.; Enam, S.; Lara, C.; Ortiz-Hidalgo, C.; Katsetos, C.D.; Khalili, K. Detection of JC Polyomavirus DNA sequences and cellular localization of T-Antigen and Agnoprotein in Oligodendrogliomas. Clin. Cancer Res. 2002, 8, 3332–3340. [Google Scholar] [PubMed]
- Gordon, J.W.; Scangos, G.A.; Plotkin, D.J.; Barbosa, J.A.; Ruddle, F.H. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 7380–7384. [Google Scholar] [CrossRef]
- Gordon, J.W.; Ruddle, F.H. Integration and stable germ line transmission of genes injected into mouse pronuclei. Science 1981, 214, 1244–1246. [Google Scholar] [CrossRef]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta NeuroPathol. 2016, 131, 803–820. [Google Scholar] [CrossRef]
- Krynska, B.; Otte, J.; Franks, R.; Khalili, K.; Croul, S. Human ubiquitous JCV(CY) T-Antigen gene induces brain tumors in experimental animals. Oncogene 1999, 18, 39–46. [Google Scholar] [CrossRef]
- Krynska, B.; Del Valle, L.; Gordon, J.; Otte, J.; Croul, S.; Khalili, K. Identification of a novel p53 mutation in JCV-induced mouse medulloblastoma. Virology 2000, 274, 65–74. [Google Scholar] [CrossRef][Green Version]
- Gordon, J.; Del Valle, L.; Otte, J.; Khalili, K. Pituitary neoplasia induced by expression of human neurotropic Polyomavirus, JCV, early genome in transgenic mice. Oncogene 2000, 19, 4840–4846. [Google Scholar] [CrossRef]
- Shollar, D.; Del Valle, L.; Khalili, K.; Otte, J.; Gordon, J. JCV T-Antigen interacts with the neurofibromatosis type 2 gene product in a transgenic mouse model of malignant peripheral nerve sheath tumors. Oncogene 2004, 23, 5459–5467. [Google Scholar] [CrossRef] [PubMed]
- White, M.K.; Khalili, K. Polyomaviruses and human cancer: Molecular mechanisms underlying patterns of tumorigenesis. Virology 2004, 324, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Dyson, N.; Bernards, R.; Friend, S.H.; Gooding, L.R.; Hassell, J.A.; Major, E.O.; Pipas, J.M.; Vandyke, T.; Harlow, E. Large T-Antigens of many Polyomaviruses are able to form complexes with the Retinoblastoma protein. J. Virol. 1990, 64, 1353–1356. [Google Scholar] [CrossRef] [PubMed]
- Levine, A.J. p53, the cellular gatekeeper for growth and division. Cell 1997, 88, 323–331. [Google Scholar] [CrossRef]
- Enam, S.; Del Valle, L.; Lara, C.; Gan, D.D.; Ortiz-Hidalgo, C.; Palazzo, J.P.; Khalili, K. Association of human Polyomavirus JCV with colon cancer: Evidence for interaction of viral T-Antigen and beta-catenin. Cancer Res. 2002, 62, 7093–7101. [Google Scholar]
- Del Valle, L.; Enam, S.; Lassak, A.; Wang, J.Y.; Croul, S.; Khalili, K.; Reiss, K. Insulin-like growth factor I receptor activity in human medulloblastomas. Clin. Cancer Res. 2002, 8, 1822–1830. [Google Scholar]
- Lassak, A.; Del Valle, L.; Peruzzi, F.; Wang, J.Y.; Enam, S.; Croul, S.; Khalili, K.; Reiss, K. Insulin receptor substrate 1 translocation to the nucleus by the human JC Virus T-Antigen. J. Biol. Chem. 2002, 277, 17231–17238. [Google Scholar] [CrossRef]
- Neel, J.V.; Major, E.O.; Awa, A.A.; Glover, T.; Burgess, A.; Traub, R.; Curfman, B.; Satoh, C. Hypothesis: “Rogue cell”-type chromosomal damage in lymphocytes is associated with infection with the JC human polyoma virus and has implications for oncopenesis. Proc. Natl. Acad. Sci. USA 1996, 93, 2690–2695. [Google Scholar] [CrossRef]
- Tognon, M.; Casalone, R.; Martini, F.; De Mattei, M.; Granata, P.; Minelli, E.; Arcuri, C.; Collini, P.; Bocchini, V. Large T-Antigen coding sequences of two DNA tumor viruses, BK and SV40, and nonrandom chromosome changes in two Glioblastoma cell lines. Cancer Genet. Cytogenet. 1996, 90, 17–23. [Google Scholar] [CrossRef]
- Trojanek, J.; Croul, S.; Ho, T.; Wang, J.Y.; Darbinyan, A.; Nowicki, M.; Del Valle, L.; Skorski, T.; Khalili, K.; Reiss, K. T-Antigen of the human Polyomavirus JC attenuates faithful DNA repair by forcing nuclear interaction between IRS-1 and Rad51. J. Cell Physiol. 2006, 206, 35–46. [Google Scholar] [CrossRef]
- Darbinyan, A.; White, M.K.; Akan, S.; Radhakrishnan, S.; Del Valle, L.; Amini, S.; Khalili, K. Alterations of DNA damage repair pathways resulting from JCV infection. Virology 2007, 364, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.; Shuda, M.; Chang, Y.; Moore, P.S. Clonal integration of a Polyomavirus in human Merkel cell carcinoma. Science 2008, 319, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Ricciardiello, L.; Chang, D.K.; Laghi, L.; Goel, A.; Chang, C.L.; Boland, C.R. Mad-1 is the exclusive JC Virus strain present in the human colon, and its transcriptional control region has a deleted 98-base-pair sequence in colon cancer tissues. J. Virol. 2001, 75, 1996–2001. [Google Scholar] [CrossRef]
- Tognon, M.; Corallini, A.; Martini, F.; Negrini, M.; Barbanti-Brodano, G. Oncogenic transformation by BK Virus and association with human tumors. Oncogene 2003, 22, 5192–5200. [Google Scholar] [CrossRef] [PubMed]
- Muller, D.C.; Ramo, M.; Naegele, K.; Ribi, S.; Wetterauer, C.; Perrina, V.; Quagliata, L.; Vlajnic, T.; Ruiz, C.; Balitzki, B.; et al. Donor-derived, metastatic urothelial cancer after kidney transplantation associated with a potentially oncogenic BK polyomavirus. J. Pathol. 2018, 244, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Y.; Sun, J.; Bao, J.; Zhu, T. BK polyomavirus infection promotes growth and aggressiveness in bladder cancer. Virol. J. 2020, 17, 139. [Google Scholar] [CrossRef]
- Krynska, B.; Gordon, J.; Otte, J.; Franks, R.; Knobler, R.; DeLuca, A.; Giordano, A.; Khalili, K. Role of cell cycle regulators in tumor formation in transgenic mice expressing the human neurotropic virus, JCV, early protein. J. Cell Biochem. 1997, 67, 223–230. [Google Scholar] [CrossRef]
- Ferreira, D.A.; Tayyar, Y.; Idris, A.; McMillan, N.A.J. A “hit-and-run” affair—A possible link for cancer progression in virally driven cancers. Biochim. Biophys. Acta Rev. Cancer 2020, 1875, 188476. [Google Scholar] [CrossRef]
- Piña-Oviedo, S.; Urbanska, K.; Radhakrishnan, S.; Sweet, T.; Reiss, K.; Khalili, K.; Del Valle, L. Effects of JC Virus infection on anti-apoptotic protein Survivin in Progressive Multifocal Leukoencephalopathy. Am. J. Pathol. 2007, 170, 1291–1304. [Google Scholar] [CrossRef]
- Del Valle, L.; Sweet, T.; Parker-Struckhoff, A.; Perez-Liz, G.; Pina-Oviedo, S. JCPyV T-Antigen activation of the anti-apoptotic Survivin promoter—Its role in the development of Progressive Multifocal Leukoencephalopathy. Viruses 2020, 12, 1253. [Google Scholar] [CrossRef]
- Ripple, M.J.; Parker Struckhoff, A.; Trillo-Tinoco, J.; Li, L.; Margolin, D.A.; McGoey, R.; Del Valle, L. Activation of c-Myc and cyclin D1 by JCV T-Antigen and beta-catenin in colon cancer. PLoS ONE 2014, 9, e106257. [Google Scholar]
- Donadoni, M.; Sariyer, R.; Wollebo, H.; Bellizzi, A.; Sariyer, I.K. Viral tumor antigen expression is no longer required in radiation-resistant subpopulation of JCV induced mouse medulloblastoma cells. Genes Cancer 2018, 9, 130–141. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dela Cruz, F.N., Jr.; Giannitti, F.; Li, L.; Woods, L.W.; Del Valle, L.; Delwart, E.; Pesavento, P.A. Novel Polyomavirus associated with Brain Tumors in Free-Ranging Raccoons, Western United States. Emerg. Infect. Dis. 2013, 19, 77–84. [Google Scholar] [CrossRef]
- Brostoff, T.; Dela Cruz, F.N., Jr.; Church, M.E.; Woolard, K.D.; Pesavento, P.A. The Raccoon Polyomavirus genome and tumor antigen transcription are stable and abundant in neuroglial tumors. J. Virol. 2014, 88, 12816–12824. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primitive Neuroectodermal Tumors and Medulloblastomas | Pituitary Tumors | Malignant Peripheral Nerve Sheath Tumors | Adrenal Neuroblastomas | Glioblastomas |
|---|---|---|---|---|
| 178 | 21 | 18 | 7 | 4 |
| PNET + Pituitary | PNET + MPNST | |||
| 5 | 7 |
| Primitive Neuroectodermal Tumors (178) | ||||||
|---|---|---|---|---|---|---|
| Spinal Cord | Brainstem | Cerebellum Medulloblastomas | Basal Ganglia | Cerebral Hemispheres | Hippocampus | Olfactory Bulb |
| 7 | 69 | 8 | 52 | 34 | 4 | 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Valle, L.; Khalili, K. Induction of Brain Tumors by the Archetype Strain of Human Neurotropic JCPyV in a Transgenic Mouse Model. Viruses 2021, 13, 162. https://doi.org/10.3390/v13020162
Del Valle L, Khalili K. Induction of Brain Tumors by the Archetype Strain of Human Neurotropic JCPyV in a Transgenic Mouse Model. Viruses. 2021; 13(2):162. https://doi.org/10.3390/v13020162
Chicago/Turabian StyleDel Valle, Luis, and Kamel Khalili. 2021. "Induction of Brain Tumors by the Archetype Strain of Human Neurotropic JCPyV in a Transgenic Mouse Model" Viruses 13, no. 2: 162. https://doi.org/10.3390/v13020162
APA StyleDel Valle, L., & Khalili, K. (2021). Induction of Brain Tumors by the Archetype Strain of Human Neurotropic JCPyV in a Transgenic Mouse Model. Viruses, 13(2), 162. https://doi.org/10.3390/v13020162