
ViralRecall—A Flexible Command-Line Tool for the Detection of Giant Virus Signatures in ‘Omic Data


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. NCDLV Genomes Used for Database Construction
2.2. Giant Virus Orthologous Groups (GVOGs)
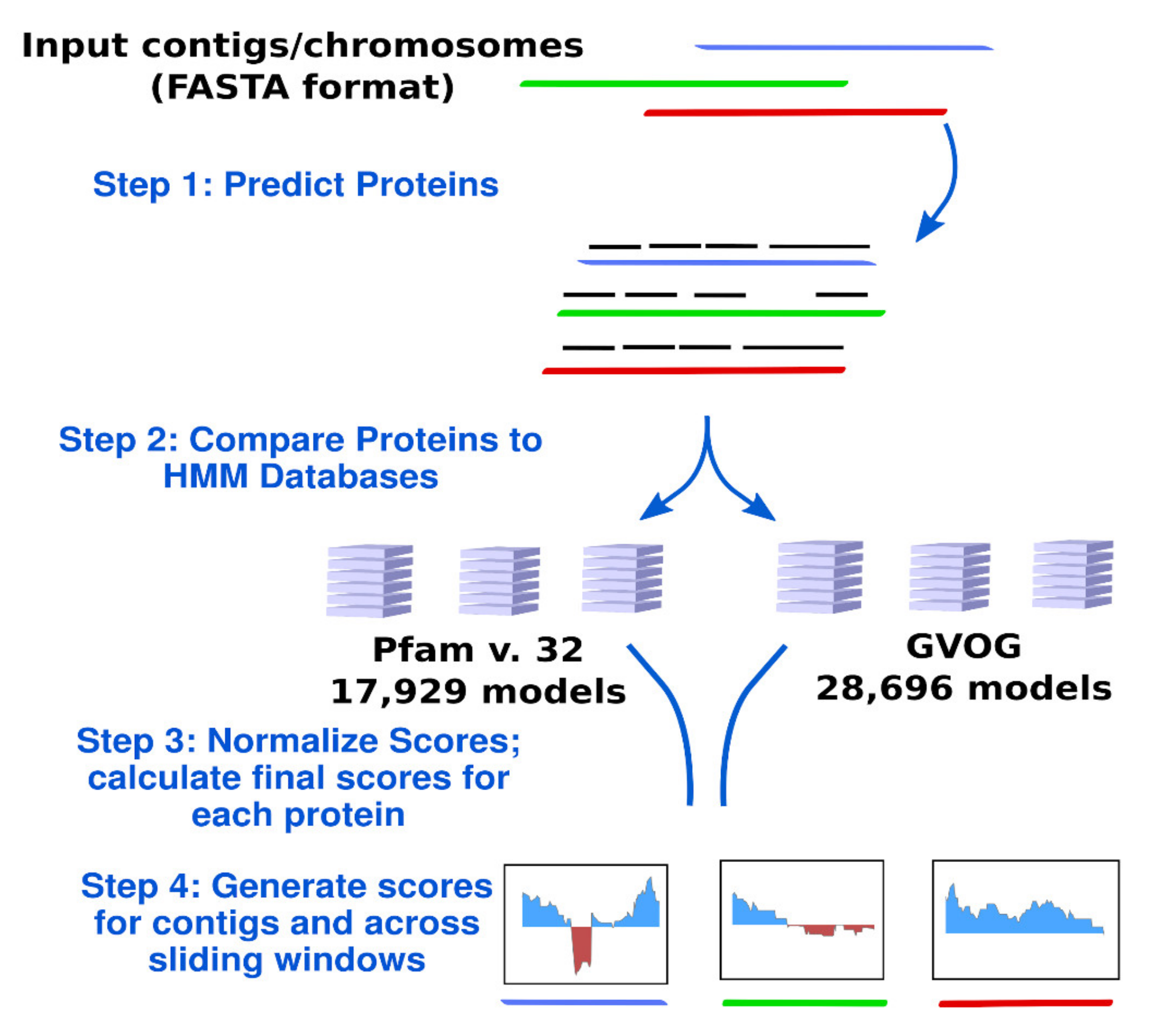
2.3. Calculation of ViralRecall Scores
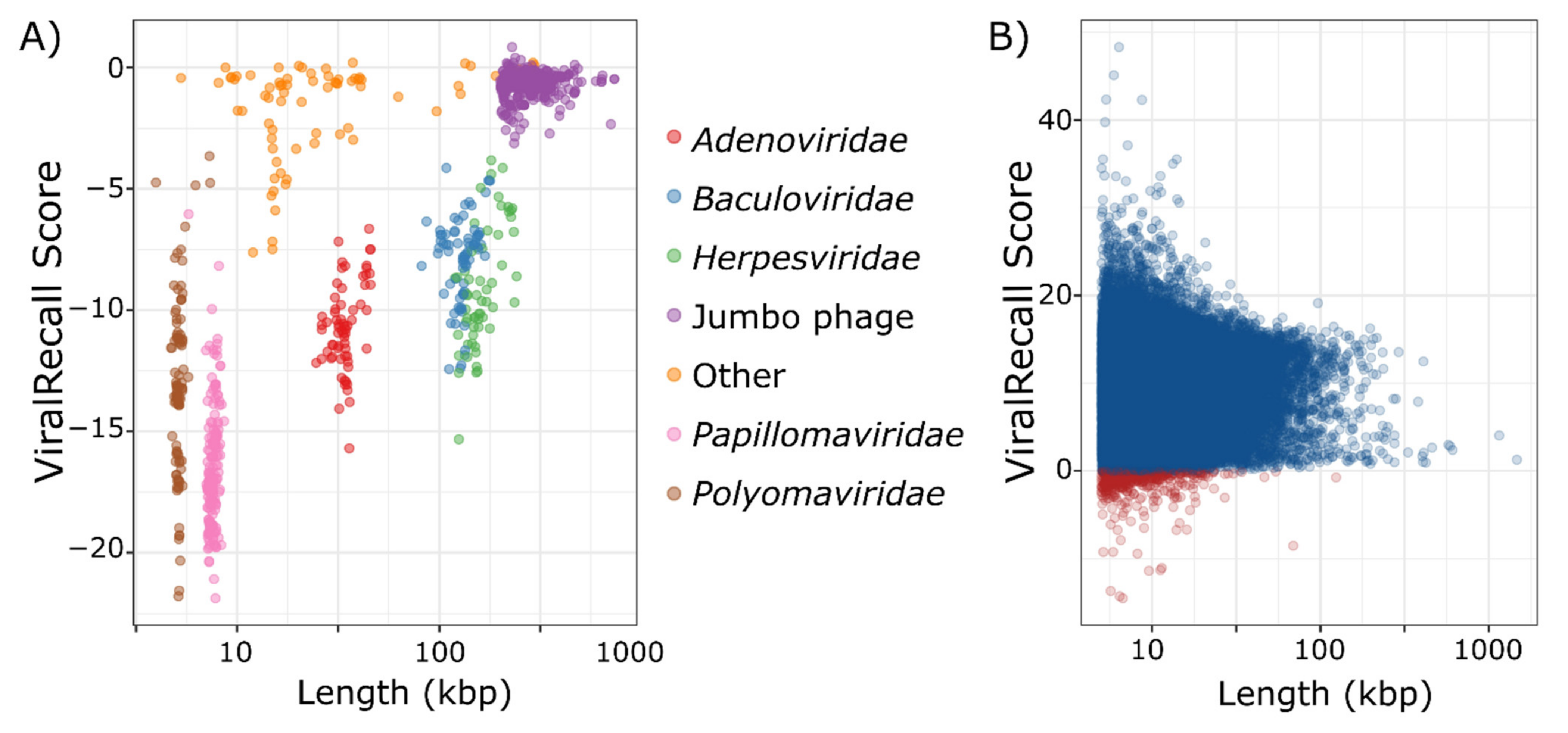
2.4. Benchmarking
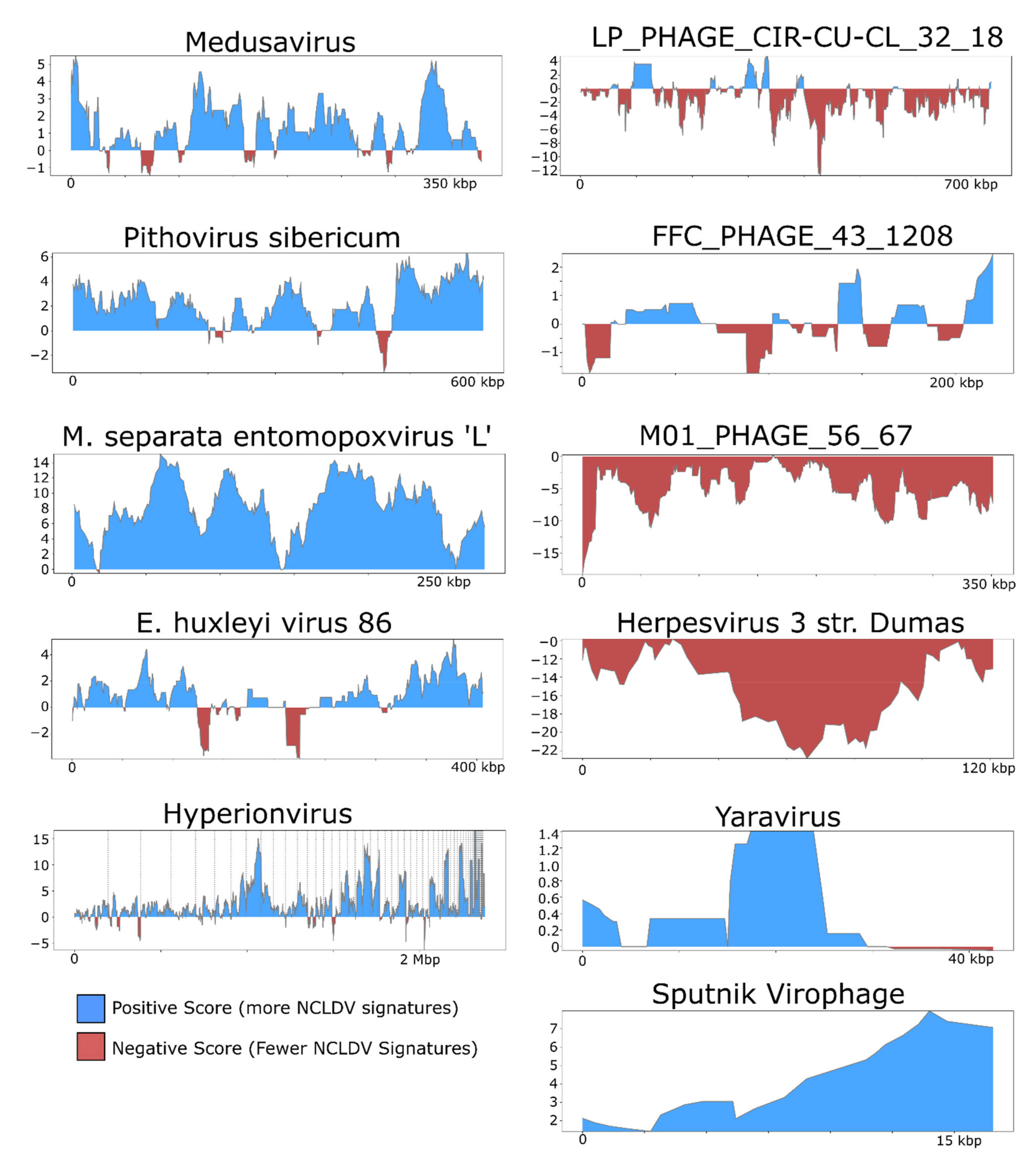
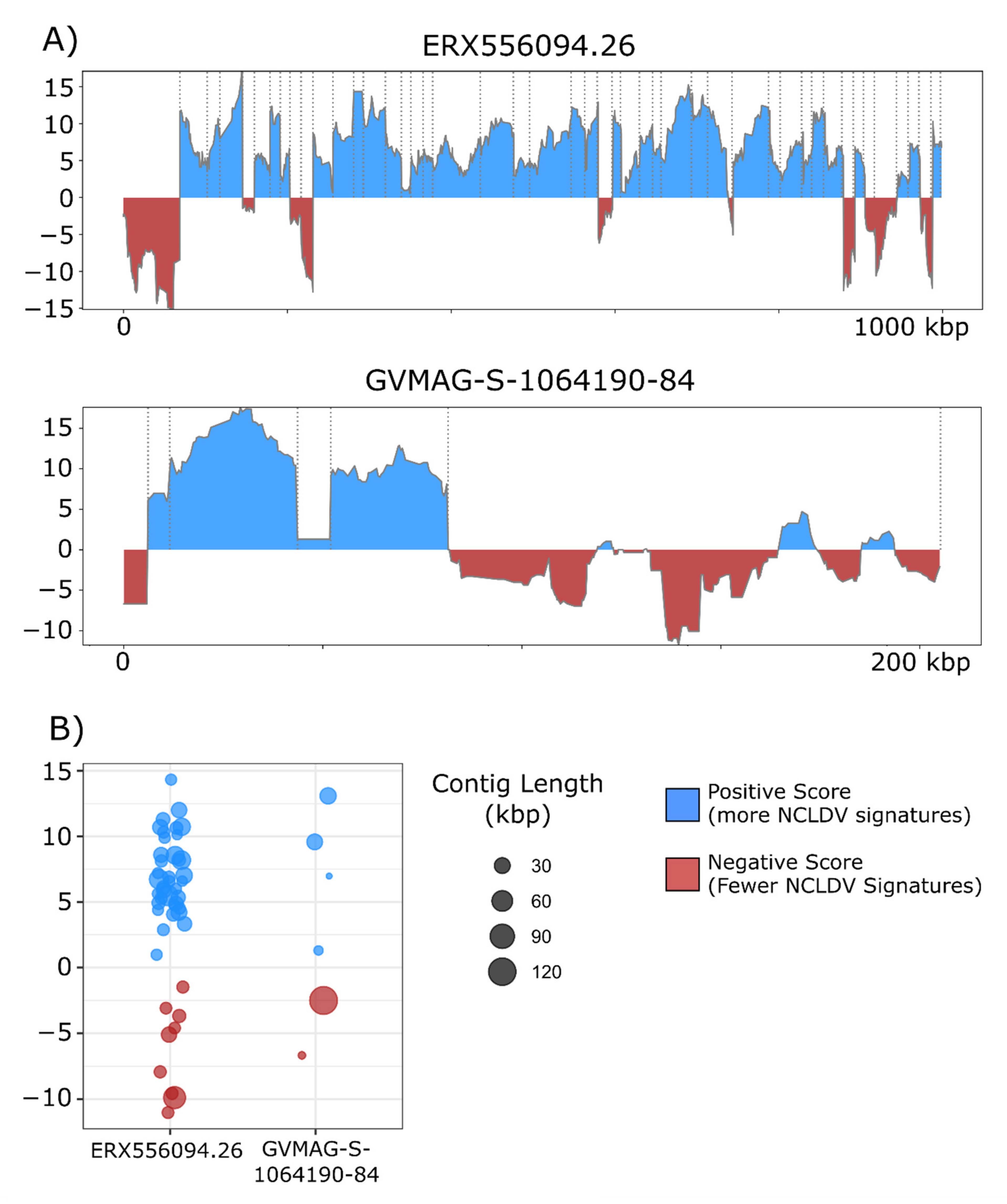
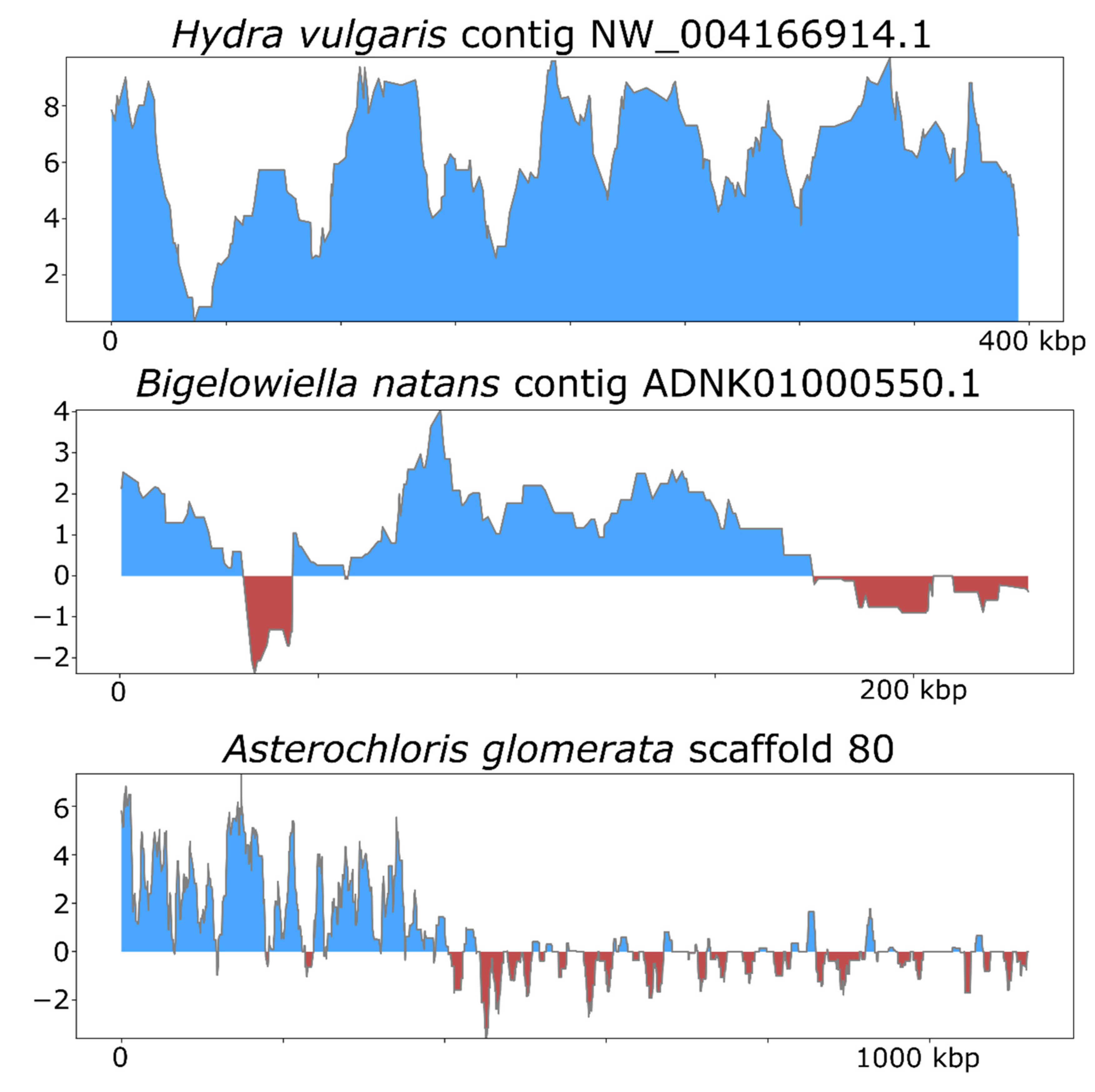
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Koonin, E.V.; Dolja, V.V.; Krupovic, M.; Varsani, A.; Wolf, Y.I.; Yutin, N.; Zerbini, F.M.; Kuhn, J.H. Global Organization and Proposed Megataxonomy of the Virus World. Microbiol. Mol. Biol. Rev. 2020, 84. [Google Scholar] [CrossRef]
- Brandes, N.; Linial, M. Giant Viruses—Big Surprises. Viruses 2019, 11, 404. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Forterre, P. Redefining viruses: Lessons from Mimivirus. Nat. Rev. Microbiol. 2008, 6, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.-W.; Yang, C.-L.; Kao, T.-T.; Wang, T.-H.; Lai, M.-W.; Ku, C. Host Range and Coding Potential of Eukaryotic Giant Viruses. Viruses 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Abergel, C.; Legendre, M.; Claverie, J.-M. The rapidly expanding universe of giant viruses: Mimivirus, Pandoravirus, Pithovirus and Mollivirus. FEMS Microbiol. Rev. 2015, 39, 779–796. [Google Scholar] [CrossRef] [PubMed]
- Aherfi, S.; Colson, P.; La Scola, B.; Raoult, D. Giant Viruses of Amoebas: An Update. Front. Microbiol. 2016, 7, 349. [Google Scholar] [CrossRef]
- Wilson, W.H.; Schroeder, D.C.; Allen, M.J.; Holden, M.T.G.; Parkhill, J.; Barrell, B.G.; Churcher, C.; Hamlin, N.; Mungall, K.; Norbertczak, H.; et al. Complete genome sequence and lytic phase transcription profile of a Coccolithovirus. Science 2005, 309, 1090–1092. [Google Scholar] [CrossRef]
- Moreau, H.; Piganeau, G.; Desdevises, Y.; Cooke, R.; Derelle, E.; Grimsley, N. Marine prasinovirus genomes show low evolutionary divergence and acquisition of protein metabolism genes by horizontal gene transfer. J. Virol. 2010, 84, 12555–12563. [Google Scholar] [CrossRef]
- Schvarcz, C.R.; Steward, G.F. A giant virus infecting green algae encodes key fermentation genes. Virology 2018, 518, 423–433. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Martinez-Gutierrez, C.A.; Weinheimer, A.R.; Aylward, F.O. Dynamic genome evolution and complex virocell metabolism of globally-distributed giant viruses. Nat. Commun. 2020, 11, 1710. [Google Scholar] [CrossRef]
- Cunha, V.D.; Da Cunha, V.; Gaia, M.; Ogata, H.; Jaillon, O.; Delmont, T.O.; Forterre, P. Giant viruses encode novel types of actins possibly related to the origin of eukaryotic actin: The viractins. bioRxiv 2020. [Google Scholar] [CrossRef]
- Abrahão, J.; Silva, L.; Silva, L.S.; Khalil, J.Y.B.; Rodrigues, R.; Arantes, T.; Assis, F.; Boratto, P.; Andrade, M.; Kroon, E.G.; et al. Tailed giant Tupanvirus possesses the most complete translational apparatus of the known virosphere. Nat. Commun. 2018, 9, 749. [Google Scholar] [CrossRef] [PubMed]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.-M. The 1.2-megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Yutin, N.; Ivanova, N.N.; Ortega, D.R.; Lee, T.K.; Vierheilig, J.; Daims, H.; Horn, M.; Wagner, M.; Jensen, G.J.; et al. Giant viruses with an expanded complement of translation system components. Science 2017, 356, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Hingamp, P.; Grimsley, N.; Acinas, S.G.; Clerissi, C.; Subirana, L.; Poulain, J.; Ferrera, I.; Sarmento, H.; Villar, E.; Lima-Mendez, G.; et al. Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes. ISME J. 2013, 7, 1678–1695. [Google Scholar] [CrossRef] [PubMed]
- Mihara, T.; Koyano, H.; Hingamp, P.; Grimsley, N.; Goto, S.; Ogata, H. Taxon Richness of “Megaviridae” Exceeds those of Bacteria and Archaea in the Ocean. Microbes Environ. 2018, 33, 162–171. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Blanc-Mathieu, R.; Li, Y.; Salazar, G.; Henry, N.; Labadie, K.; de Vargas, C.; Sullivan, M.B.; Bowler, C.; Wincker, P.; et al. Biogeography of marine giant viruses reveals their interplay with eukaryotes and ecological functions. Nat Ecol Evol 2020, 4, 1639–1649. [Google Scholar] [CrossRef]
- Moniruzzaman, M.; Weinheimer, A.R.; Martinez-Gutierrez, C.A.; Aylward, F.O. Widespread endogenization of giant viruses shapes genomes of green algae. Nature 2020, 588, 141–145. [Google Scholar] [CrossRef]
- Filée, J. Multiple occurrences of giant virus core genes acquired by eukaryotic genomes: The visible part of the iceberg? Virology 2014, 466-467, 53–59. [Google Scholar] [CrossRef]
- Gallot-Lavallée, L.; Blanc, G. A Glimpse of Nucleo-Cytoplasmic Large DNA Virus Biodiversity through the Eukaryotic Genomics Window. Viruses 2017, 9. [Google Scholar] [CrossRef]
- Lang, D.; Ullrich, K.K.; Murat, F.; Fuchs, J.; Jenkins, J.; Haas, F.B.; Piednoel, M.; Gundlach, H.; Van Bel, M.; Meyberg, R.; et al. The Physcomitrella patens chromosome-scale assembly reveals moss genome structure and evolution. Plant J. 2018, 93, 515–533. [Google Scholar] [CrossRef] [PubMed]
- Maumus, F.; Epert, A.; Nogué, F.; Blanc, G. Plant genomes enclose footprints of past infections by giant virus relatives. Nat. Commun. 2014, 5, 4268. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Roux, S.; Paez-Espino, D.; Jungbluth, S.; Walsh, D.A.; Denef, V.J.; McMahon, K.D.; Konstantinidis, K.T.; Eloe-Fadrosh, E.A.; Kyrpides, N.C.; et al. Giant virus diversity and host interactions through global metagenomics. Nature 2020, 578, 432–436. [Google Scholar] [CrossRef] [PubMed]
- Bäckström, D.; Yutin, N.; Jørgensen, S.L.; Dharamshi, J.; Homa, F.; Zaremba-Niedwiedzka, K.; Spang, A.; Wolf, Y.I.; Koonin, E.V.; Ettema, T.J.G. Virus Genomes from Deep Sea Sediments Expand the Ocean Megavirome and Support Independent Origins of Viral Gigantism. MBio 2019, 10. [Google Scholar] [CrossRef]
- Verneau, J.; Levasseur, A.; Raoult, D.; La Scola, B.; Colson, P. MG-Digger: An Automated Pipeline to Search for Giant Virus-Related Sequences in Metagenomes. Front. Microbiol. 2016, 7, 428. [Google Scholar] [CrossRef]
- Kerepesi, C.; Grolmusz, V. The “Giant Virus Finder” discovers an abundance of giant viruses in the Antarctic dry valleys. Arch. Virol. 2017, 162, 1671–1676. [Google Scholar] [CrossRef]
- Tithi, S.S.; Aylward, F.O.; Jensen, R.V.; Zhang, L. FastViromeExplorer: A pipeline for virus and phage identification and abundance profiling in metagenomics data. PeerJ 2018, 6, e4227. [Google Scholar] [CrossRef]
- Boyer, M.; Yutin, N.; Pagnier, I.; Barrassi, L.; Fournous, G.; Espinosa, L.; Robert, C.; Azza, S.; Sun, S.; Rossmann, M.G.; et al. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc. Natl. Acad. Sci. USA 2009, 106, 21848–21853. [Google Scholar] [CrossRef]
- VOGDB Virus Orthologous Groups. Available online: https://vogdb.csb.univie.ac.at/ (accessed on 11 January 2021).
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: Current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef]
- Ondov, B.D.; Treangen, T.J.; Melsted, P.; Mallonee, A.B.; Bergman, N.H.; Koren, S.; Phillippy, A.M. Mash: Fast genome and metagenome distance estimation using MinHash. Genome Biol. 2016, 17, 132. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.; Findeiss, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics 2011, 12, 124. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLoS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Söding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Capella-Gutiérrez, S.; Silla-Martínez, J.M.; Gabaldón, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef]
- Huerta-Cepas, J.; Szklarczyk, D.; Heller, D.; Hernández-Plaza, A.; Forslund, S.K.; Cook, H.; Mende, D.R.; Letunic, I.; Rattei, T.; Jensen, L.J.; et al. eggNOG 5.0: A hierarchical, functionally and phylogenetically annotated orthology resource based on 5090 organisms and 2502 viruses. Nucleic Acids Res. 2019, 47, D309–D314. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses ICTV. Available online: https://talk.ictvonline.org (accessed on 1 November 2020).
- Al-Shayeb, B.; Sachdeva, R.; Chen, L.-X.; Ward, F.; Munk, P.; Devoto, A.; Castelle, C.J.; Olm, M.R.; Bouma-Gregson, K.; Amano, Y.; et al. Clades of huge phages from across Earth’s ecosystems. Nature 2020, 578, 425–431. [Google Scholar] [CrossRef]
- La Scola, B.; Desnues, C.; Pagnier, I.; Robert, C.; Barrassi, L.; Fournous, G.; Merchat, M.; Suzan-Monti, M.; Forterre, P.; Koonin, E.; et al. The virophage as a unique parasite of the giant mimivirus. Nature 2008, 455, 100–104. [Google Scholar] [CrossRef]
- GenomeTools. Available online: http://genometools.org/ (accessed on 1 December 2020).
- Yoshikawa, G.; Blanc-Mathieu, R.; Song, C.; Kayama, Y.; Mochizuki, T.; Murata, K.; Ogata, H.; Takemura, M. Medusavirus, a Novel Large DNA Virus Discovered from Hot Spring Water. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 4274–4279. [Google Scholar] [CrossRef] [PubMed]
- Thézé, J.; Takatsuka, J.; Li, Z.; Gallais, J.; Doucet, D.; Arif, B.; Nakai, M.; Herniou, E.A. New insights into the evolution of Entomopoxvirinae from the complete genome sequences of four entomopoxviruses infecting Adoxophyes honmai, Choristoneura biennis, Choristoneura rosaceana, and Mythimna separata. J. Virol. 2013, 87, 7992–8003. [Google Scholar] [CrossRef] [PubMed]
- Schulz, F.; Alteio, L.; Goudeau, D.; Ryan, E.M.; Yu, F.B.; Malmstrom, R.R.; Blanchard, J.; Woyke, T. Hidden diversity of soil giant viruses. Nat. Commun. 2018, 9, 4881. [Google Scholar] [CrossRef] [PubMed]
- Davison, A.J.; Scott, J.E. The Complete DNA Sequence of Varicella-Zoster Virus. Journal of General Virology 1986, 67, 1759–1816. [Google Scholar] [CrossRef]
- Boratto, P.V.M.; Oliveira, G.P.; Machado, T.B.; Andrade, A.C.S.P.; Baudoin, J.-P.; Klose, T.; Schulz, F.; Azza, S.; Decloquement, P.; Chabrière, E.; et al. Yaravirus: A novel 80-nm virus infecting. Proc. Natl. Acad. Sci. USA 2020, 117, 16579–16586. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Blanc, G.; Gallot-Lavallée, L.; Maumus, F. Provirophages in the Bigelowiella genome bear testimony to past encounters with giant viruses. Proc. Natl. Acad. Sci. USA 2015, 112, E5318–E5326. [Google Scholar] [CrossRef] [PubMed]
- Armaleo, D.; Müller, O.; Lutzoni, F.; Andrésson, Ó.S.; Blanc, G.; Bode, H.B.; Collart, F.R.; Dal Grande, F.; Dietrich, F.; Grigoriev, I.V.; et al. The lichen symbiosis re-viewed through the genomes of Cladonia grayi and its algal partner Asterochloris glomerata. BMC Genomics 2019, 20, 605. [Google Scholar] [CrossRef] [PubMed]
- Weinheimer, A.R.; Aylward, F.O. A distinct lineage of Caudovirales that encodes a deeply branching multi-subunit RNA polymerase. Nat. Commun. 2020, 11, 4506. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aylward, F.O.; Moniruzzaman, M. ViralRecall—A Flexible Command-Line Tool for the Detection of Giant Virus Signatures in ‘Omic Data. Viruses 2021, 13, 150. https://doi.org/10.3390/v13020150
Aylward FO, Moniruzzaman M. ViralRecall—A Flexible Command-Line Tool for the Detection of Giant Virus Signatures in ‘Omic Data. Viruses. 2021; 13(2):150. https://doi.org/10.3390/v13020150
Chicago/Turabian StyleAylward, Frank O., and Mohammad Moniruzzaman. 2021. "ViralRecall—A Flexible Command-Line Tool for the Detection of Giant Virus Signatures in ‘Omic Data" Viruses 13, no. 2: 150. https://doi.org/10.3390/v13020150
APA StyleAylward, F. O., & Moniruzzaman, M. (2021). ViralRecall—A Flexible Command-Line Tool for the Detection of Giant Virus Signatures in ‘Omic Data. Viruses, 13(2), 150. https://doi.org/10.3390/v13020150