
Molecular Basis of Epstein–Barr Virus Latency Establishment and Lytic Reactivation

 , , and
, , and
Abstract
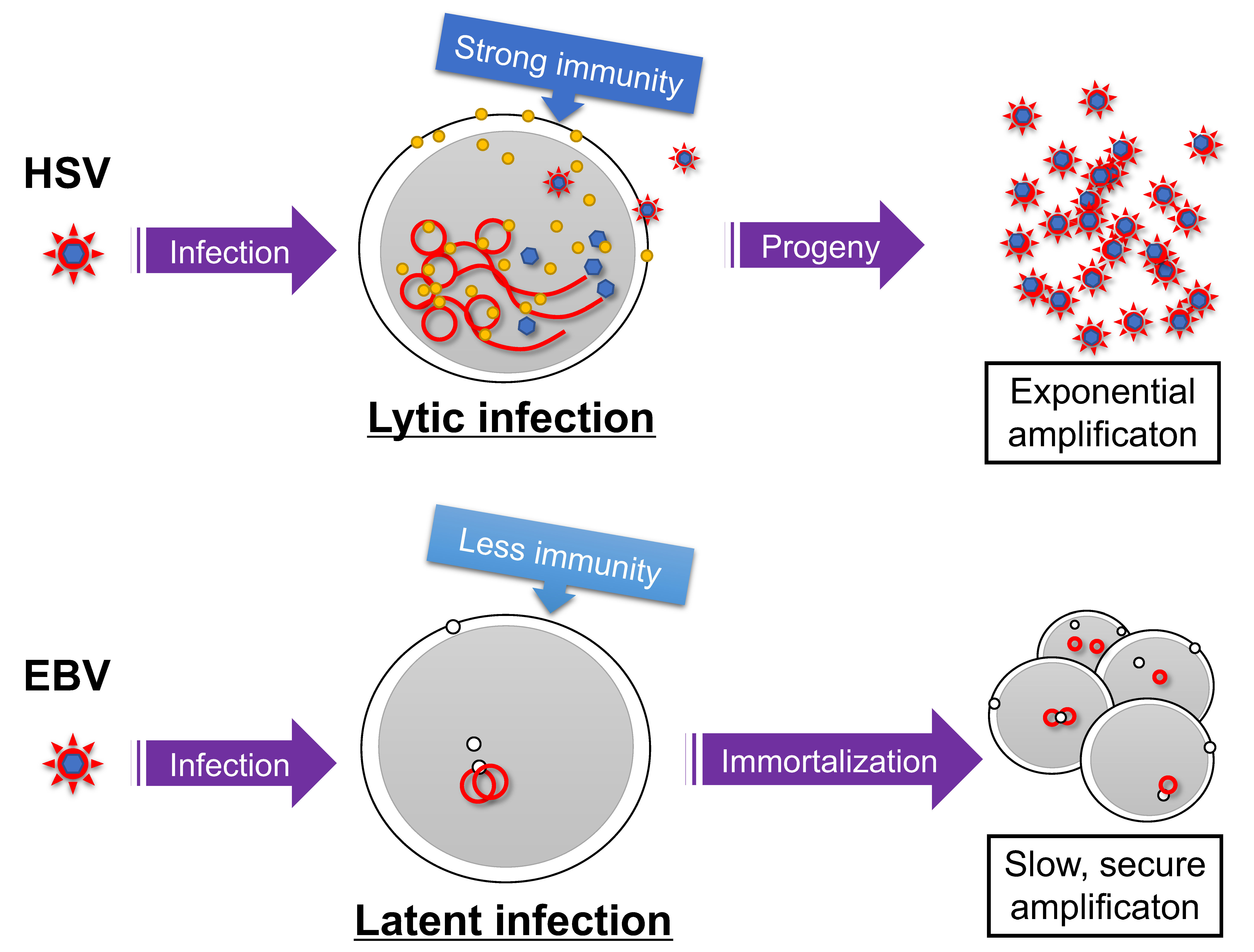
1. Introduction
2. Establishment of Latency
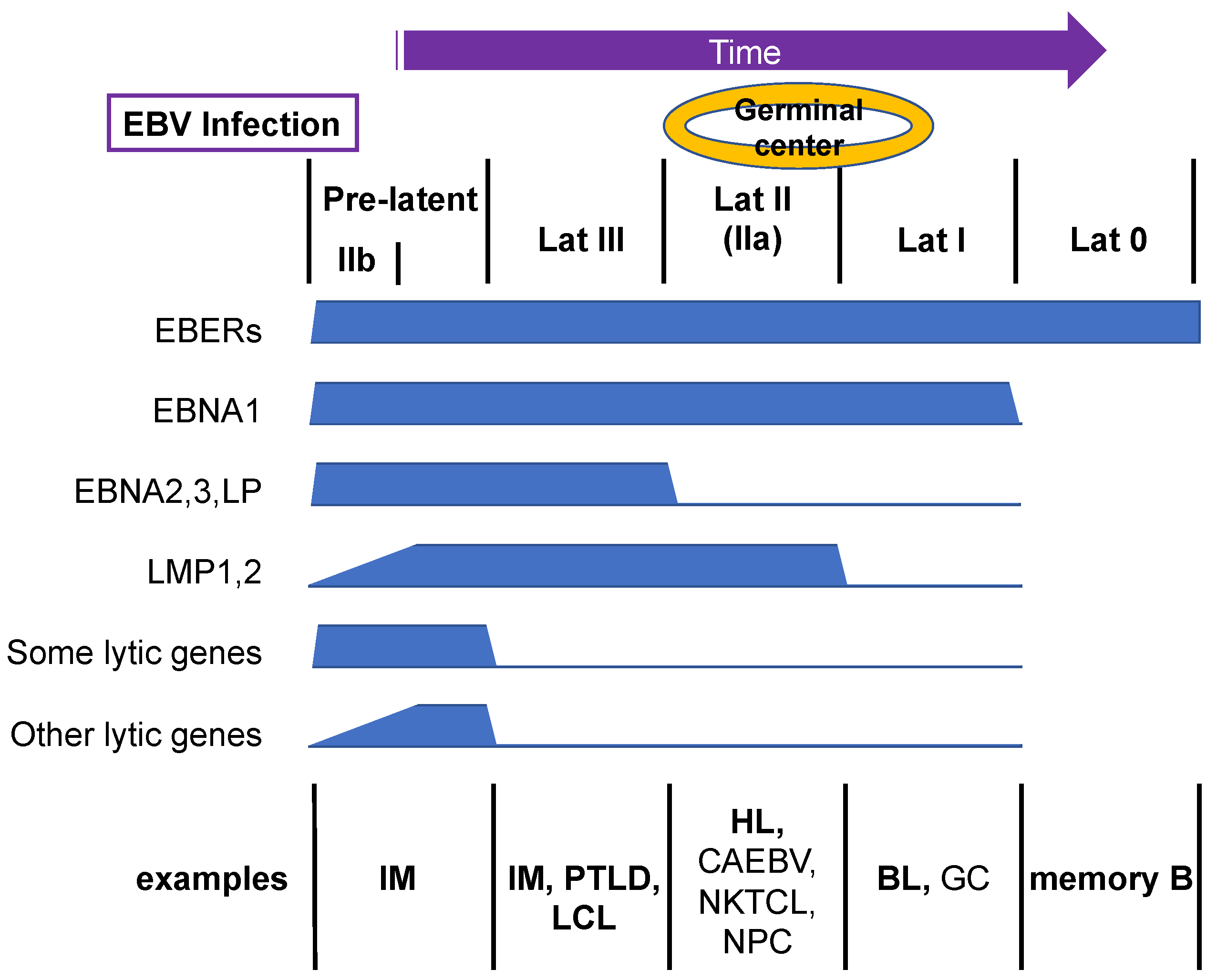
3. Restriction of Latent Genes
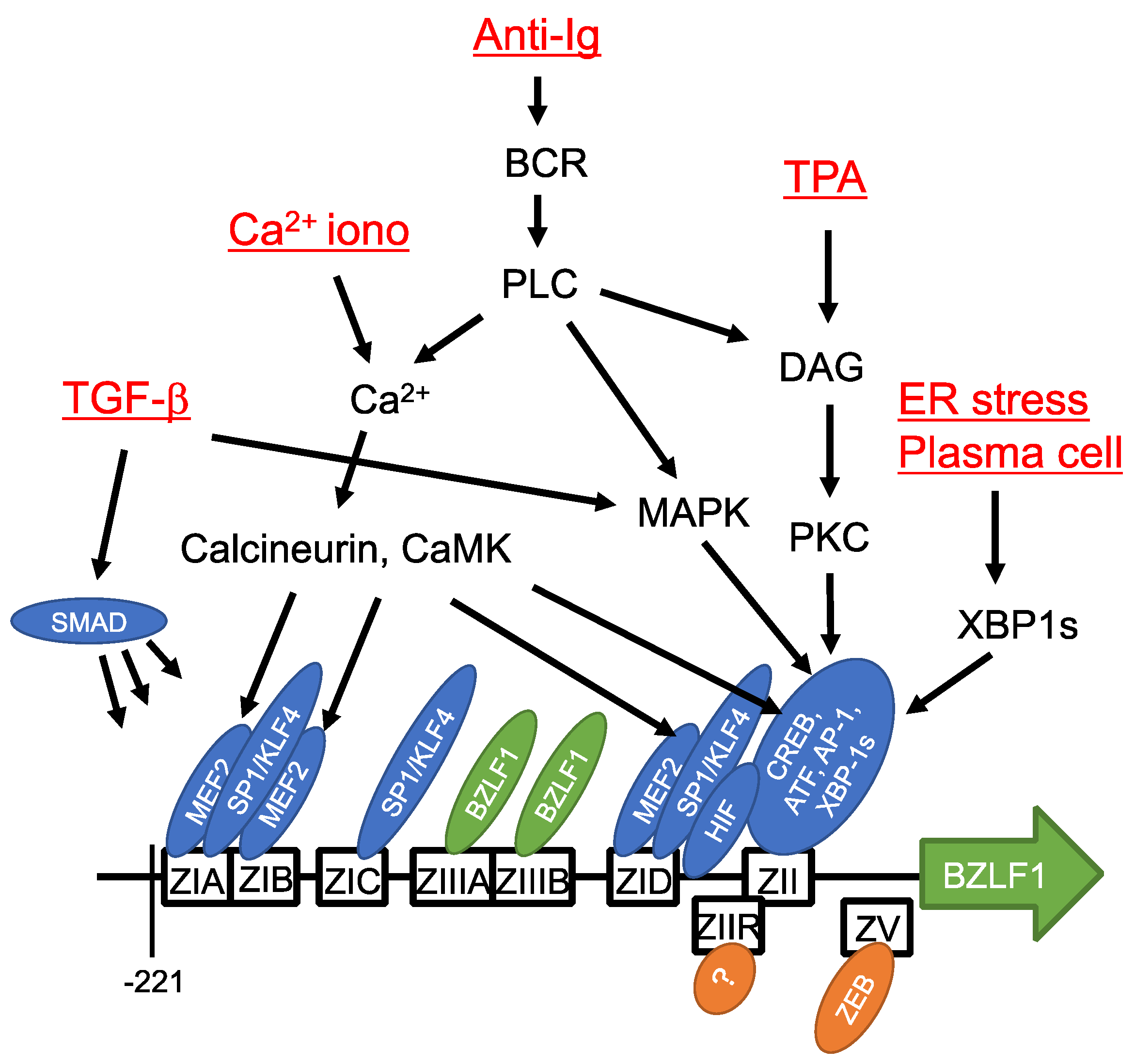
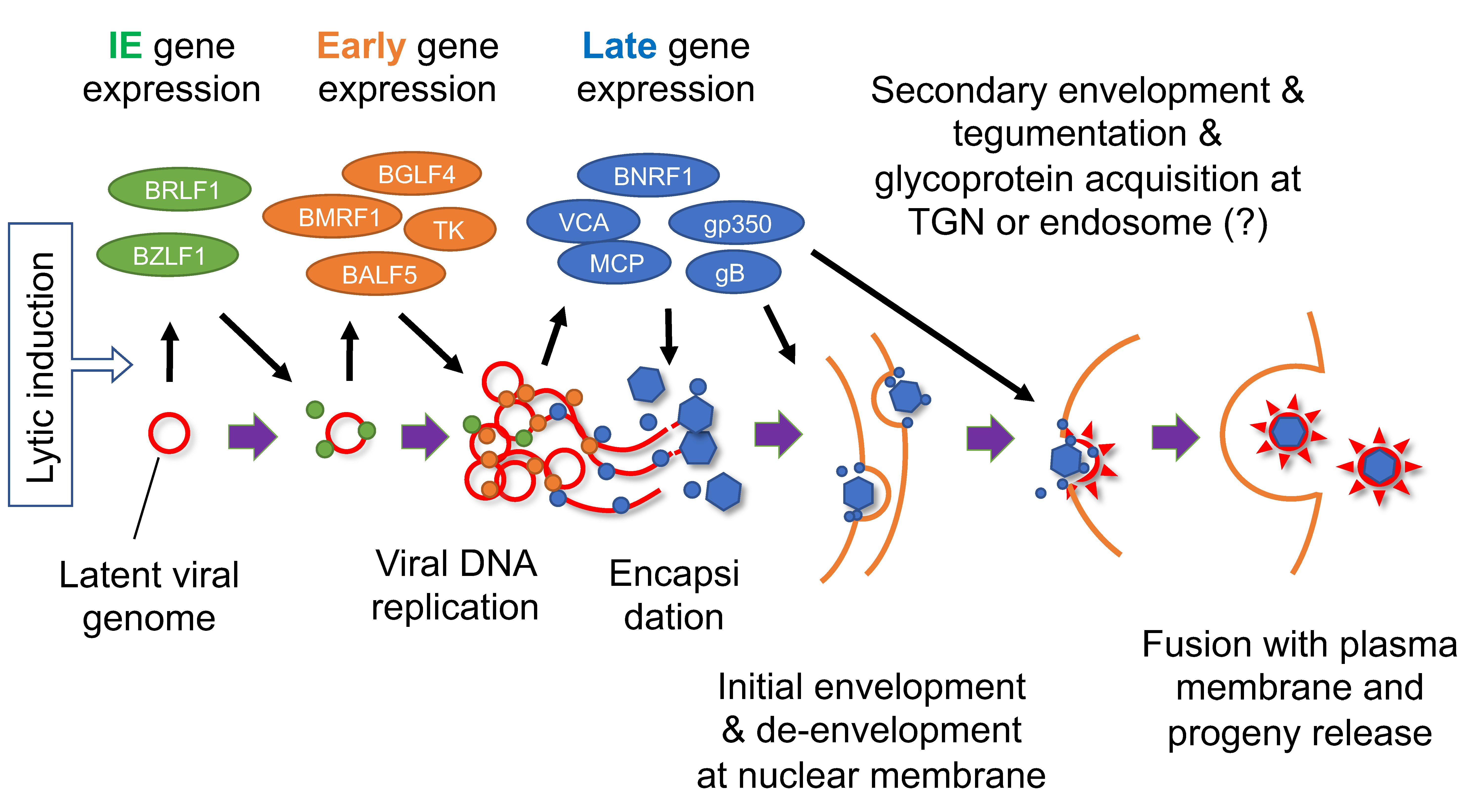
4. Reactivation from Latency
5. Epigenetic Regulation of Latency Establishment
6. Epigenetic Regulation of Latent Gene Restriction
7. Epigenetic Regulation of Reactivation
8. Involvement of Lytic Genes in Oncogenesis
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Young, L.S.; Yap, L.-F.; Murray, P.G. Epstein–Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Dunmire, S.K.; Hogquist, K.A.; Balfour, H.H. Infectious Mononucleosis. Curr. Top. Microbiol. Immunol. 2015, 390, 211–240. [Google Scholar] [PubMed]
- Cohen, J.I. Epstein-Barr virus infection. N. Engl. J. Med. 2000, 343, 481–492. [Google Scholar] [CrossRef]
- Kenney, S.C. Reactivation and lytic replication of EBV. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.I. Herpesvirus latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390, 151–209. [Google Scholar] [PubMed]
- Wille, C.K.; Nawandar, D.M.; Panfil, A.R.; Ko, M.M.; Hagemeier, S.R.; Kenney, S.C. Viral Genome Methylation Differentially Affects the Ability of BZLF1 versus BRLF1 To Activate Epstein-Barr Virus Lytic Gene Expression and Viral Replication. J. Virol. 2013, 87, 935–950. [Google Scholar] [CrossRef]
- Temple, R.M.; Zhu, J.; Budgeon, L.; Christensen, N.D.; Meyers, C.; Sample, C.E. Efficient replication of Epstein–Barr virus in stratified epithelium in vitro. Proc. Natl. Acad. Sci. USA 2014, 111, 16544–16549. [Google Scholar] [CrossRef]
- Yoshiyama, H.; Shimizu, N.; Takada, K. Persistent Epstein-Barr virus infection in a human T-cell line: Unique program of latent virus expression. EMBO J. 1995, 14, 3706–3711. [Google Scholar] [CrossRef]
- Murata, T.; Sato, Y.; Kimura, H. Modes of infection and oncogenesis by the Epstein-Barr virus. Rev. Med Virol. 2014, 24, 242–253. [Google Scholar] [CrossRef]
- Kalla, M.; Göbel, C.; Hammerschmidt, W. The Lytic Phase of Epstein-Barr Virus Requires a Viral Genome with 5-Methylcytosine Residues in CpG Sites. J. Virol. 2012, 86, 447–458. [Google Scholar] [CrossRef]
- Inagaki, T.; Sato, Y.; Ito, J.; Takaki, M.; Okuno, Y.; Yaguchi, M.; Al Masud, H.M.A.; Watanabe, T.; Sato, K.; Iwami, S.; et al. Direct Evidence of Abortive Lytic Infection-Mediated Establishment of Epstein-Barr Virus Latency During B-Cell Infection. Front. Microbiol. 2021, 11, 575255. [Google Scholar] [CrossRef]
- Rosemarie, Q.; Sugden, B. Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef] [PubMed]
- Altmann, M.; Hammerschmidt, W. Epstein-Barr Virus Provides a New Paradigm: A Requirement for the Immediate Inhibition of Apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar] [CrossRef]
- Quinn, L.L.; Williams, L.R.; White, C.; Forrest, C.; Zuo, J.; Rowe, M. The Missing Link in Epstein-Barr Virus Immune Evasion: The BDLF3 Gene Induces Ubiquitination and Downregulation of Major Histocompatibility Complex Class I (MHC-I) and MHC-II. J. Virol. 2016, 90, 356–367. [Google Scholar] [CrossRef] [PubMed]
- Jochum, S.; Ruiss, R.; Moosmann, A.; Hammerschmidt, W.; Zeidler, R. RNAs in Epstein-Barr virions control early steps of infection. Proc. Natl. Acad. Sci. USA 2012, 109, E1396–E1404. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Luftig, M.A. Dynamic Epstein-Barr virus gene expression on the path to B-cell transformation. Adv. Virus Res. 2014, 88, 279–313. [Google Scholar] [CrossRef]
- Wang, C.; Li, D.; Zhang, L.; Jiang, S.; Liang, J.; Narita, Y.; Hou, I.; Zhong, Q.; Zheng, Z.; Xiao, H.; et al. RNA Sequencing Analyses of Gene Expression during Epstein-Barr Virus Infection of Primary B Lymphocytes. J. Virol. 2019, 93, e00226-19. [Google Scholar] [CrossRef] [PubMed]
- Kanda, T. EBV-Encoded Latent Genes. Adv. Exp. Med. Biol. 2018, 1045, 377–394. [Google Scholar] [CrossRef]
- Kempkes, B.; Ling, P.D. EBNA2 and Its Coactivator EBNA-LP. Curr. Top. Microbiol. Immunol. 2015, 391, 35–59. [Google Scholar] [CrossRef]
- Contreras-Brodin, B.; Karlsson, A.; Nilsson, T.; Rymo, L.; Klein, G. B cell-specific activation of the Epstein--Barr virus-encoded C promoter compared with the wide-range activation of the W promoter. J. Gen. Virol. 1996, 77, 1159–1162. [Google Scholar] [CrossRef] [PubMed]
- Kirby, H.; Rickinson, A.; Bell, A. The activity of the Epstein–Barr virus BamHI W promoter in B cells is dependent on the binding of CREB/ATF factors. J. Gen. Virol. 2000, 81, 1057–1066. [Google Scholar] [CrossRef]
- Tierney, R.; Kirby, H.; Nagra, J.; Rickinson, A.; Bell, A. The Epstein-Barr Virus Promoter Initiating B-Cell Transformation Is Activated by RFX Proteins and the B-Cell-Specific Activator Protein BSAP/Pax5. J. Virol. 2000, 74, 10458–10467. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Tierney, R.; Nagra, J.; Hutchings, I.; Shannon-Lowe, C.; Altmann, M.; Hammerschmidt, W.; Rickinson, A.; Bell, A. Epstein-Barr Virus Exploits BSAP/Pax5 To Achieve the B-Cell Specificity of Its Growth-Transforming Program. J. Virol. 2007, 81, 10092–10100. [Google Scholar] [CrossRef] [PubMed]
- Tierney, R.J.; Kao, K.Y.; Nagra, J.K.; Rickinson, A.B. Epstein-Barr virus BamHI W repeat number limits EBNA2/EBNA-LP coexpression in newly infected B cells and the efficiency of B-cell transformation: A rationale for the multiple W repeats in wild-type virus strains. J. Virol. 2011, 85, 12362–12375. [Google Scholar] [CrossRef]
- Woisetschlaeger, M.; Jin, X.W.; Yandava, C.N.; Furmanski, L.A.; Strominger, J.L.; Speck, S.H. Role for the Epstein-Barr virus nuclear antigen 2 in viral promoter switching during initial stages of infection. Proc. Natl. Acad. Sci. USA 1991, 88, 3942–3946. [Google Scholar] [CrossRef] [PubMed]
- Mabuchi, S.; Hijioka, F.; Watanabe, T.; Yanagi, Y.; Okuno, Y.; Al Masud, H.M.A.; Sato, Y.; Murata, T.; Kimura, H. Role of Epstein–Barr Virus C Promoter Deletion in Diffuse Large B Cell Lymphoma. Cancers 2021, 13, 561. [Google Scholar] [CrossRef]
- Borestrom, C.; Forsman, A.; Ruetschi, U.; Rymo, L. E2F1, ARID3A/Bright and Oct-2 factors bind to the Epstein-Barr virus C promoter, EBNA1 and oriP, participating in long-distance promoter-enhancer interactions. J. Gen. Virol. 2012, 93, 1065–1075. [Google Scholar] [CrossRef] [PubMed]
- Boreström, C.; Zetterberg, H.; Liff, K.; Rymo, L. Functional Interaction of Nuclear Factor Y and Sp1 Is Required for Activation of the Epstein-Barr Virus C Promoter. J. Virol. 2003, 77, 821–829. [Google Scholar] [CrossRef][Green Version]
- Price, A.M.; Luftig, M.A. To Be or Not IIb: A Multi-Step Process for Epstein-Barr Virus Latency Establishment and Consequences for B Cell Tumorigenesis. PLoS Pathog. 2015, 11, e1004656. [Google Scholar] [CrossRef]
- Yoshida, M.; Murata, T.; Ashio, K.; Narita, Y.; Watanabe, T.; Al Masud, H.M.A.; Sato, Y.; Goshima, F.; Kimura, H. Characterization of a Suppressive Cis-acting Element in the Epstein–Barr Virus LMP1 Promoter. Front. Microbiol. 2017, 8, 2302. [Google Scholar] [CrossRef]
- Price, A.M.; Messinger, J.E.; Luftig, M.A. c-Myc Represses Transcription of Epstein-Barr Virus Latent Membrane Protein 1 Early after Primary B Cell Infection. J. Virol. 2018, 92, e01178-17. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Tsang, S.F.; Kurilla, M.G.; Cohen, J.I.; Kieff, E. Epstein-Barr virus nuclear antigen 2 transactivates latent membrane protein LMP1. J. Virol. 1990, 64, 3407–3416. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Grossman, S.R.; Kieff, E. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 430–435. [Google Scholar] [CrossRef]
- Demetriades, C.; Mosialos, G. The LMP1 promoter can be transactivated directly by NF-kappaB. J. Virol. 2009, 83, 5269–5277. [Google Scholar] [CrossRef] [PubMed]
- Johansson, P.; Jansson, A.; Ruetschi, U.; Rymo, L. Nuclear factor-kappaB binds to the Epstein-Barr Virus LMP1 promoter and upregulates its expression. J. Virol. 2009, 83, 1393–1401. [Google Scholar] [CrossRef]
- Murata, T.; Noda, C.; Narita, Y.; Watanabe, T.; Yoshida, M.; Ashio, K.; Sato, Y.; Goshima, F.; Kanda, T.; Yoshiyama, H.; et al. Induction of Epstein-Barr Virus Oncoprotein LMP1 by Transcription Factors AP-2 and Early B Cell Factor. J. Virol. 2016, 90, 3873–3889. [Google Scholar] [CrossRef]
- Ning, S.; Hahn, A.M.; Huye, L.E.; Pagano, J.S. Interferon Regulatory Factor 7 Regulates Expression of Epstein-Barr Virus Latent Membrane Protein 1: A Regulatory Circuit. J. Virol. 2003, 77, 9359–9368. [Google Scholar] [CrossRef] [PubMed]
- Sjoblom, A.; Yang, W.; Palmqvist, L.; Jansson, A.; Rymo, L. An ATF/CRE element mediates both EBNA2-dependent and EBNA2-independent activation of the Epstein-Barr virus LMP1 gene promoter. J. Virol. 1998, 72, 1365–1376. [Google Scholar] [CrossRef]
- Wang, L.W.; Jiang, S.; Gewurz, B.E. Epstein-Barr Virus LMP1-Mediated Oncogenicity. J. Virol. 2017, 91, e01718-16. [Google Scholar] [CrossRef] [PubMed]
- Yanagi, Y.; Okuno, Y.; Narita, Y.; Al Masud, H.A.; Watanabe, T.; Sato, Y.; Kanda, T.; Kimura, H.; Murata, T. RNAseq analysis identifies involvement of EBNA2 in PD-L1 induction during Epstein-Barr virus infection of primary B cells. Virology 2021, 557, 44–54. [Google Scholar] [CrossRef]
- Ling, P.D.; Hsieh, J.J.; Ruf, I.K.; Rawlins, D.R.; Hayward, S.D. EBNA-2 upregulation of Epstein-Barr virus latency promoters and the cellular CD23 promoter utilizes a common targeting intermediate, CBF1. J. Virol. 1994, 68, 5375–5383. [Google Scholar] [CrossRef]
- Boccellato, F.; Anastasiadou, E.; Rosato, P.; Kempkes, B.; Frati, L.; Faggioni, A. EBNA2 interferes with the germinal center phenotype by downregulating BCL6 and TCL1 in non-Hodgkin’s lymphoma cells. J. Virol. 2007, 81, 2274–2282. [Google Scholar] [CrossRef] [PubMed]
- Kis, L.L.; Salamon, D.; Persson, E.K.; Nagy, N.; Scheeren, F.; Spits, H.; Klein, G.; Klein, E. IL-21 imposes a type II EBV gene expression on type III and type I B cells by the repression of C- and activation of LMP-1-promoter. Proc. Natl. Acad. Sci. USA 2009, 107, 872–877. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Hutt-Fletcher, L.; Cao, L.; Hayward, S.D. A Positive Autoregulatory Loop of LMP1 Expression and STAT Activation in Epithelial Cells Latently Infected with Epstein-Barr Virus. J. Virol. 2003, 77, 4139–4148. [Google Scholar] [CrossRef]
- Kis, L.L.; Gerasimčik, N.; Salamon, D.; Persson, E.K.; Nagy, N.; Klein, G.; Severinson, E.; Klein, E. STAT6 signaling pathway activated by the cytokines IL-4 and IL-13 induces expression of the Epstein-Barr virus–encoded protein LMP-1 in absence of EBNA-2: Implications for the type II EBV latent gene expression in Hodgkin lymphoma. Blood 2011, 117, 165–174. [Google Scholar] [CrossRef]
- Kis, L.L.; Takahara, M.; Nagy, N.; Klein, G.; Klein, E. Cytokine mediated induction of the major Epstein–Barr virus (EBV)-encoded transforming protein, LMP-1. Immunol. Lett. 2006, 104, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Kis, L.L.; Takahara, M.; Nagy, N.; Klein, G.; Klein, E. IL-10 can induce the expression of EBV-encoded latent membrane protein-1 (LMP-1) in the absence of EBNA-2 in B lymphocytes and in Burkitt lymphoma- and NK lymphoma-derived cell lines. Blood 2006, 107, 2928–2935. [Google Scholar] [CrossRef]
- Chen, H.; Lee, J.M.; Zong, Y.; Borowitz, M.; Ng, M.H.; Ambinder, R.F.; Hayward, S.D. Linkage between STAT Regulation and Epstein-Barr Virus Gene Expression in Tumors. J. Virol. 2001, 75, 2929–2937. [Google Scholar] [CrossRef]
- Noda, C.; Murata, T.; Kanda, T.; Yoshiyama, H.; Sugimoto, A.; Kawashima, D.; Saito, S.; Isomura, H.; Tsurumi, T. Identification and Characterization of CCAAT Enhancer-binding Protein (C/EBP) as a Transcriptional Activator for Epstein-Barr Virus Oncogene Latent Membrane Protein 1. J. Biol. Chem. 2011, 286, 42524–42533. [Google Scholar] [CrossRef]
- Kenney, S.C.; Mertz, J.E. Regulation of the latent-lytic switch in Epstein–Barr virus. Semin. Cancer Biol. 2014, 26, 60–68. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, J.; El-Guindy, A. Epstein-Barr Virus Lytic Cycle Reactivation. Curr. Top. Microbiol. Immunol. 2015, 391, 237–261. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Tsurumi, T. Switching of EBV cycles between latent and lytic states. Rev. Med. Virol. 2014, 24, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.; El-Guindy, A.; Countryman, J.; Ye, J.; Gradoville, L. Lytic Cycle Switches of Oncogenic Human Gammaherpesviruses. Adv. Cancer Res. 2007, 97, 81–109. [Google Scholar] [CrossRef] [PubMed]
- Sinclair, A.J. bZIP proteins of human gammaherpesviruses. J. Gen. Virol. 2003, 84, 1941–1949. [Google Scholar] [CrossRef]
- Stolz, M.L.; McCormick, C. The bZIP Proteins of Oncogenic Viruses. Viruses 2020, 12, 757. [Google Scholar] [CrossRef]
- Packham, G.; Economou, A.; Rooney, C.M.; Rowe, D.T.; Farrell, P.J. Structure and function of the Epstein-Barr virus BZLF1 protein. J. Virol. 1990, 64, 2110–2116. [Google Scholar] [CrossRef]
- Lieberman, P.M.; Hardwick, J.M.; Sample, J.; Hayward, G.S.; Hayward, S.D. The zta transactivator involved in induction of lytic cycle gene expression in Epstein-Barr virus-infected lymphocytes binds to both AP-1 and ZRE sites in target promoter and enhancer regions. J. Virol. 1990, 64, 1143–1155. [Google Scholar] [CrossRef]
- Urier, G.; Buisson, M.; Chambard, P.; Sergeant, A. The Epstein-Barr virus early protein EB1 activates transcription from different responsive elements including AP-1 binding sites. EMBO J. 1989, 8, 1447–1453. [Google Scholar] [CrossRef]
- Bhende, P.M.; Seaman, W.T.; Delecluse, H.-J.; Kenney, S.C. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat. Genet. 2004, 36, 1099–1104. [Google Scholar] [CrossRef]
- Dickerson, S.J.; Xing, Y.; Robinson, A.R.; Seaman, W.T.; Gruffat, H.; Kenney, S.C. Methylation-Dependent Binding of the Epstein-Barr Virus BZLF1 Protein to Viral Promoters. PLoS Pathog. 2009, 5, e1000356. [Google Scholar] [CrossRef] [PubMed]
- Woellmer, A.; Arteaga-Salas, J.M.; Hammerschmidt, W. BZLF1 governs CpG-methylated chromatin of Epstein-Barr Virus reversing epigenetic repression. PLoS Pathog. 2012, 8, e1002902. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, T.; Fujita, M.; Kudoh, A. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med Virol. 2005, 15, 3–15. [Google Scholar] [CrossRef] [PubMed]
- Gruffat, H.; Duran, N.; Buisson, M.; Wild, F.; Buckland, R.; Sergeant, A. Characterization of an R-binding site mediating the R-induced activation of the Epstein-Barr virus BMLF1 promoter. J. Virol. 1992, 66, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Manet, E.; Rigolet, A.; Gruffat, H.; Giot, J.-F.; Sergeant, A. Domains of the Epstein-Barr virus (EBV) transcription factor R required for dimerization, DNA binding and activation. Nucleic Acids Res. 1991, 19, 2661–2667. [Google Scholar] [CrossRef][Green Version]
- Chang, L.-K.; Chung, J.-Y.; Hong, Y.-R.; Ichimura, T.; Nakao, M.; Liu, S.-T. Activation of Sp1-mediated transcription by Rta of Epstein-Barr virus via an interaction with MCAF1. Nucleic Acids Res. 2005, 33, 6528–6539. [Google Scholar] [CrossRef] [PubMed]
- Robinson, A.R.; Kwek, S.S.; Hagemeier, S.R.; Wille, C.K.; Kenney, S.C. Cellular transcription factor Oct-1 interacts with the Epstein-Barr virus BRLF1 protein to promote disruption of viral latency. J. Virol. 2011, 85, 8940–8953. [Google Scholar] [CrossRef][Green Version]
- Darr, C.D.; Mauser, A.; Kenney, S. Epstein-Barr Virus Immediate-Early Protein BRLF1 Induces the Lytic Form of Viral Replication through a Mechanism Involving Phosphatidylinositol-3 Kinase Activation. J. Virol. 2001, 75, 6135–6142. [Google Scholar] [CrossRef]
- Lee, Y.-H.; Chiu, Y.-F.; Wang, W.-H.; Chang, L.-K.; Liu, S.-T. Activation of the ERK signal transduction pathway by Epstein–Barr virus immediate-early protein Rta. J. Gen. Virol. 2008, 89, 2437–2446. [Google Scholar] [CrossRef]
- Flemington, E.; Speck, S.H. Identification of phorbol ester response elements in the promoter of Epstein-Barr virus putative lytic switch gene BZLF1. J. Virol. 1990, 64, 1217–1226. [Google Scholar] [CrossRef]
- Daibata, M.; Speck, S.H.; Mulder, C.; Sairenji, T. Regulation of the BZLF1 Promoter of Epstein-Barr Virus by Second Messengers in Anti-immunoglobulin-Treated B Cells. Virology 1994, 198, 446–454. [Google Scholar] [CrossRef]
- Liu, S.; Borras, A.M.; Liu, P.; Suske, G.; Speck, S.H. Binding of the Ubiquitous Cellular Transcription Factors Sp1 and Sp3 to the ZI Domains in the Epstein–Barr Virus Lytic Switch BZLF1 Gene Promoter. Virology 1997, 228, 11–18. [Google Scholar] [CrossRef]
- Liu, S.; Liu, P.; Borras, A.; Chatila, T.; Speck, S.H. Cyclosporin A-sensitive induction of the Epstein-Barr virus lytic switch is mediated via a novel pathway involving a MEF2 family member. EMBO J. 1997, 16, 143–153. [Google Scholar] [CrossRef]
- Murata, T.; Narita, Y.; Sugimoto, A.; Kawashima, D.; Kanda, T.; Tsurumi, T.; Li, M.-T.; Di, W.; Xu, H.; Yang, Y.-K.; et al. Contribution of Myocyte Enhancer Factor 2 Family Transcription Factors to BZLF1 Expression in Epstein-Barr Virus Reactivation from Latency. J. Virol. 2013, 87, 10148–10162. [Google Scholar] [CrossRef]
- Nawandar, D.M.; Wang, A.; Makielski, K.; Lee, D.; Ma, S.; Barlow, E.; Reusch, J.; Jiang, R.; Wille, C.K.; Greenspan, D.; et al. Differentiation-Dependent KLF4 Expression Promotes Lytic Epstein-Barr Virus Infection in Epithelial Cells. PLoS Pathog. 2015, 11, e1005195. [Google Scholar] [CrossRef]
- Sun, C.C.; Thorley-Lawson, D.A. Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J. Virol. 2007, 81, 13566–13577. [Google Scholar] [CrossRef]
- Wang, Y.-C.J.; Huang, J.-M.; Montalvo, E.A. Characterization of Proteins Binding to the ZII Element in the Epstein–Barr Virus BZLF1 Promoter: Transactivation by ATF1. Virology 1997, 227, 323–330. [Google Scholar] [CrossRef][Green Version]
- Flemington, E.; Speck, S.H. Autoregulation of Epstein-Barr virus putative lytic switch gene BZLF1. J. Virol. 1990, 64, 1227–1232. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.J.; Cordes, B.-L.A.; Sathiamoorthi, S.; Patel, P.; Yuan, X.; Iempridee, T.; Yu, X.; Lee, D.L.; Lambert, P.F.; Mertz, J.E. Reactivation of Epstein-Barr Virus by HIF-1α Requires p53. J. Virol. 2020, 94, e00722-20. [Google Scholar] [CrossRef]
- Iempridee, T.; Das, S.; Xu, I.; Mertz, J.E. Transforming growth factor beta-induced reactivation of Epstein-Barr virus involves multiple Smad-binding elements cooperatively activating expression of the latent-lytic switch BZLF1 gene. J. Virol. 2011, 85, 7836–7848. [Google Scholar] [CrossRef] [PubMed]
- Kraus, R.J.; Perrigoue, J.G.; Mertz, J.E. ZEB Negatively Regulates the Lytic-Switch BZLF1 Gene Promoter of Epstein-Barr Virus. J. Virol. 2003, 77, 199–207. [Google Scholar] [CrossRef]
- Yu, X.; Wang, Z.; Mertz, J.E. ZEB1 Regulates the Latent-Lytic Switch in Infection by Epstein-Barr Virus. PLoS Pathog. 2007, 3, e194. [Google Scholar] [CrossRef]
- Liu, P.; Liu, S.; Speck, S.H. Identification of a negative cis element within the ZII domain of the Epstein-Barr virus lytic switch BZLF1 gene promoter. J. Virol. 1998, 72, 8230–8239. [Google Scholar] [CrossRef]
- Yu, X.; McCarthy, P.J.; Lim, H.-J.; Iempridee, T.; Kraus, R.J.; Gorlen, D.A.; Mertz, J.E. The ZIIR Element of the Epstein-Barr Virus BZLF1 Promoter Plays a Central Role in Establishment and Maintenance of Viral Latency. J. Virol. 2011, 85, 5081–5090. [Google Scholar] [CrossRef][Green Version]
- Guo, R.; Jiang, C.; Zhang, Y.; Govande, A.; Trudeau, S.J.; Chen, F.; Fry, C.J.; Puri, R.; Wolinsky, E.; Schineller, M.; et al. MYC Controls the Epstein-Barr Virus Lytic Switch. Mol. Cell 2020, 78, 653–669. [Google Scholar] [CrossRef]
- Lv, D.-W.; Zhang, K.; Li, R. Interferon regulatory factor 8 regulates caspase-1 expression to facilitate Epstein-Barr virus reactivation in response to B cell receptor stimulation and chemical induction. PLoS Pathog. 2018, 14, e1006868. [Google Scholar] [CrossRef]
- Glaser, G.; Vogel, M.; Wolf, H.; Niller, H.H. Regulation of the Epstein-Barr viral immediate early BRLF1 promoter through a distal NF1 site. Arch. Virol. 1998, 143, 1967–1983. [Google Scholar] [CrossRef]
- Reusch, J.A.; Nawandar, D.M.; Wright, K.L.; Kenney, S.C.; Mertz, J.E. Cellular Differentiation Regulator BLIMP1 Induces Epstein-Barr Virus Lytic Reactivation in Epithelial and B Cells by Activating Transcription from both the R and Z Promoters. J. Virol. 2015, 89, 1731–1743. [Google Scholar] [CrossRef]
- Sarkar, R.; Verma, S.C. Egr-1 regulates RTA transcription through a cooperative involvement of transcriptional regulators. Oncotarget 2017, 8, 91425–91444. [Google Scholar] [CrossRef]
- Zalani, S.; Coppage, A.; Holley-Guthrie, E.; Kenney, S. The cellular YY1 transcription factor binds a cis-acting, negatively regulating element in the Epstein-Barr virus BRLF1 promoter. J. Virol. 1997, 71, 3268–3274. [Google Scholar] [CrossRef]
- Zalani, S.; Holley-Guthrie, E.A.; Gutsch, D.E.; Kenney, S.C. The Epstein-Barr virus immediate-early promoter BRLF1 can be activated by the cellular Sp1 transcription factor. J. Virol. 1992, 66, 7282–7292. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Marendy, E.; Wang, Y.D.; Yuan, J.; Sample, J.T.; Swaminathan, S. Multiple roles of Epstein-Barr virus SM protein in lytic replication. J. Virol. 2007, 81, 4058–4069. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Juillard, F.; Bazot, Q.; Mure, F.; Tafforeau, L.; Macri, C.; Rabourdin-Combe, C.; Lotteau, V.; Manet, E.; Gruffat, H. Epstein–Barr virus protein EB2 stimulates cytoplasmic mRNA accumulation by counteracting the deleterious effects of SRp20 on viral mRNAs. Nucleic Acids Res. 2012, 40, 6834–6849. [Google Scholar] [CrossRef]
- Key, S.C.; Yoshizaki, T.; Pagano, J.S. The Epstein-Barr virus (EBV) SM protein enhances pre-mRNA processing of the EBV DNA polymerase transcript. J. Virol. 1998, 72, 8485–8492. [Google Scholar] [CrossRef]
- Nicewonger, J.; Suck, G.; Bloch, D.; Swaminathan, S. Epstein-Barr Virus (EBV) SM Protein Induces and Recruits Cellular Sp110b To Stabilize mRNAs and Enhance EBV Lytic Gene Expression. J. Virol. 2004, 78, 9412–9422. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ruvolo, V.; Gupta, A.K.; Swaminathan, S. Epstein-Barr Virus SM Protein Interacts with mRNA In Vivo and Mediates a Gene-Specific Increase in Cytoplasmic mRNA. J. Virol. 2001, 75, 6033–6041. [Google Scholar] [CrossRef]
- Verma, D.; Bais, S.; Gaillard, M.; Swaminathan, S. Epstein-Barr Virus SM Protein Utilizes Cellular Splicing Factor SRp20 To Mediate Alternative Splicing. J. Virol. 2010, 84, 11781–11789. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Church, T.M.; Swaminathan, S. Epstein–Barr virus co-opts TFIIH component XPB to specifically activate essential viral lytic promoters. Proc. Natl. Acad. Sci. USA 2020, 117, 13044–13055. [Google Scholar] [CrossRef] [PubMed]
- Verma, D.; Swaminathan, S. Epstein-Barr Virus SM Protein Functions as an Alternative Splicing Factor. J. Virol. 2008, 82, 7180–7188. [Google Scholar] [CrossRef]
- Fixman, E.D.; Hayward, G.S.; Hayward, S.D. Replication of Epstein-Barr virus oriLyt: Lack of a dedicated virally encoded origin-binding protein and dependence on Zta in cotransfection assays. J. Virol. 1995, 69, 2998–3006. [Google Scholar] [CrossRef]
- Su, M.-T.; Liu, I.-H.; Wu, C.-W.; Chang, S.-M.; Tsai, C.-H.; Yang, P.-W.; Chuang, Y.-C.; Lee, C.-P.; Chen, M.-R. Uracil DNA Glycosylase BKRF3 Contributes to Epstein-Barr Virus DNA Replication through Physical Interactions with Proteins in Viral DNA Replication Complex. J. Virol. 2014, 88, 8883–8899. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Schepers, A.; Pich, D.; Hammerschmidt, W. Activation of oriLyt, the lytic origin of DNA replication of Epstein-Barr virus, by BZLF1. Virology 1996, 220, 367–376. [Google Scholar] [CrossRef]
- Chakravorty, A.; Sugden, B.; Johannsen, E.C. An Epigenetic Journey: Epstein-Barr Virus Transcribes Chromatinized and Subsequently Unchromatinized Templates during Its Lytic Cycle. J. Virol. 2019, 93, e02247-18. [Google Scholar] [CrossRef] [PubMed]
- Aubry, V.; Mure, F.; Mariamé, B.; Deschamps, T.; Wyrwicz, L.; Manet, E.; Gruffat, H. Epstein-Barr Virus Late Gene Transcription Depends on the Assembly of a Virus-Specific Preinitiation Complex. J. Virol. 2014, 88, 12825–12838. [Google Scholar] [CrossRef]
- Gruffat, H.; Kadjouf, F.; Mariamé, B.; Manet, E. The Epstein-Barr Virus BcRF1 Gene Product Is a TBP-Like Protein with an Essential Role in Late Gene Expression. J. Virol. 2012, 86, 6023–6032. [Google Scholar] [CrossRef]
- Gruffat, H.; Marchione, R.; Manet, E. Herpesvirus Late Gene Expression: A Viral-Specific Pre-initiation Complex Is Key. Front. Microbiol. 2016, 7, 869. [Google Scholar] [CrossRef]
- Watanabe, T.; Narita, Y.; Yoshida, M.; Sato, Y.; Goshima, F.; Kimura, H.; Murata, T. The Epstein-Barr Virus BDLF4 Gene Is Required for Efficient Expression of Viral Late Lytic Genes. J. Virol. 2015, 89, 10120–10124. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Walsh, A.; Lam, T.T.; Delecluse, H.-J.; El-Guindy, A. A single phosphoacceptor residue in BGLF3 is essential for transcription of Epstein-Barr virus late genes. PLoS Pathog. 2019, 15, e1007980. [Google Scholar] [CrossRef]
- Sato, Y.; Watanabe, T.; Suzuki, C.; Abe, Y.; Al Masud, H.M.A.; Inagaki, T.; Yoshida, M.; Suzuki, T.; Goshima, F.; Adachi, J.; et al. S-Like-Phase Cyclin-Dependent Kinases Stabilize the Epstein-Barr Virus BDLF4 Protein to Temporally Control Late Gene Transcription. J. Virol. 2019, 93, e01707-18. [Google Scholar] [CrossRef] [PubMed]
- Murata, T. Regulation of Epstein-Barr virus reactivation from latency. Microbiol. Immunol. 2014, 58, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.-P.; Chen, M.-R. Conquering the Nuclear Envelope Barriers by EBV Lytic Replication. Viruses 2021, 13, 702. [Google Scholar] [CrossRef]
- Murata, T. Encyclopedia of EBV-Encoded Lytic Genes: An Update. Adv. Exp. Med. Biol. 2018, 1045, 395–412. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.F.; Rosales, C.; Nieva, P.L.; Graña, O.; Ballestar, E.; Ropero, S.; Espada, J.; Melo, S.; Lujambio, A.; Fraga, M.; et al. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 2009, 19, 438–451. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, E.; Luftig, M.; Chase, M.R.; Weicksel, S.; Cahir-McFarland, E.; Illanes, D.; Sarracino, D.; Kieff, E. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 2004, 101, 16286–16291. [Google Scholar] [CrossRef]
- Kalla, M.; Schmeinck, A.; Bergbauer, M.; Pich, D.; Hammerschmidt, W. AP-1 homolog BZLF1 of Epstein-Barr virus has two essential functions dependent on the epigenetic state of the viral genome. Proc. Natl. Acad. Sci. USA 2009, 107, 850–855. [Google Scholar] [CrossRef]
- Ichikawa, T.; Okuno, Y.; Sato, Y.; Goshima, F.; Yoshiyama, H.; Kanda, T.; Kimura, H.; Murata, T. Regulation of Epstein-Barr Virus Life Cycle and Cell Proliferation by Histone H3K27 Methyltransferase EZH2 in Akata Cells. mSphere 2018, 3, e00478-18. [Google Scholar] [CrossRef]
- Tsai, K.; Chan, L.; Gibeault, R.; Conn, K.; Dheekollu, J.; Domsic, J.; Marmorstein, R.; Schang, L.; Lieberman, P.M. Viral Reprogramming of the Daxx Histone H3.3 Chaperone during Early Epstein-Barr Virus Infection. J. Virol. 2014, 88, 14350–14363. [Google Scholar] [CrossRef]
- Alinari, L.; Mahasenan, K.V.; Yan, F.; Karkhanis, V.; Chung, J.-H.; Smith, E.M.; Quinion, C.; Smith, P.L.; Kim, L.; Patton, J.T.; et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood 2015, 125, 2530–2543. [Google Scholar] [CrossRef]
- Leonard, S.; Gordon, N.; Smith, N.; Rowe, M.; Murray, P.G.; Woodman, C.B. Arginine Methyltransferases Are Regulated by Epstein-Barr Virus in B Cells and Are Differentially Expressed in Hodgkin’s Lymphoma. Pathogens 2012, 1, 52–64. [Google Scholar] [CrossRef]
- Liu, C.-D.; Cheng, C.-P.; Fang, J.-S.; Chen, L.-C.; Zhao, B.; Kieff, E.; Peng, C.-W. Modulation of Epstein–Barr Virus Nuclear Antigen 2-dependent transcription by protein arginine methyltransferase 5. Biochem. Biophys. Res. Commun. 2013, 430, 1097–1102. [Google Scholar] [CrossRef][Green Version]
- Shire, K.; Kapoor, P.; Jiang, K.; Hing, M.N.T.; Sivachandran, N.; Nguyen, T.; Frappier, L. Regulation of the EBNA1 Epstein-Barr Virus Protein by Serine Phosphorylation and Arginine Methylation. J. Virol. 2006, 80, 5261–5272. [Google Scholar] [CrossRef]
- Hansen, K.D.; Sabunciyan, S.; Langmead, B.; Nagy, N.; Curley, R.; Klein, G.; Klein, E.; Salamon, D.; Feinberg, A.P. Large-scale hypomethylated blocks associated with Epstein-Barr virus-induced B-cell immortalization. Genome Res. 2014, 24, 177–184. [Google Scholar] [CrossRef]
- Hernando, H.; Islam, A.B.; Rodriguez-Ubreva, J.; Forné, I.; Ciudad, L.; Imhof, A.; Shannon-Lowe, C.; Ballestar, E. Epstein–Barr virus-mediated transformation of B cells induces global chromatin changes independent to the acquisition of proliferation. Nucleic Acids Res. 2014, 42, 249–263. [Google Scholar] [CrossRef]
- Hernando, H.; Shannon-Lowe, C.; Islam, A.B.; Al-Shahrour, F.; Rodriguez-Ubreva, J.; Cortez, V.C.R.; Javierre, B.M.; Mangas, C.; Fernández, A.F.; Parra, M.; et al. The B cell transcription program mediates hypomethylation and overexpression of key genes in Epstein-Barr virus-associated proliferative conversion. Genome Biol. 2013, 14, R3. [Google Scholar] [CrossRef]
- Stanland, L.J.; Luftig, M.A. The Role of EBV-Induced Hypermethylation in Gastric Cancer Tumorigenesis. Viruses 2020, 12, 1222. [Google Scholar] [CrossRef]
- Kaneda, A.; Matsusaka, K.; Aburatani, H.; Fukayama, M. Epstein-Barr virus infection as an epigenetic driver of tumorigenesis. Cancer Res. 2012, 72, 3445–3450. [Google Scholar] [CrossRef]
- Pott, S.; Lieb, J.D. What are super-enhancers? Nat. Genet. 2015, 47, 8–12. [Google Scholar] [CrossRef]
- Jiang, S.; Zhou, H.; Liang, J.; Gerdt, C.; Wang, C.; Ke, L.; Schmidt, S.C.; Narita, Y.; Ma, Y.; Wang, S.; et al. The Epstein-Barr Virus Regulome in Lymphoblastoid Cells. Cell Host Microbe 2017, 22, 561–573. [Google Scholar] [CrossRef]
- Zhou, H.; Schmidt, S.C.; Jiang, S.; Willox, B.; Bernhardt, K.; Liang, J.; Johannsen, E.C.; Kharchenko, P.; Gewurz, B.E.; Kieff, E.; et al. Epstein-Barr Virus Oncoprotein Super-enhancers Control B Cell Growth. Cell Host Microbe 2015, 17, 205–216. [Google Scholar] [CrossRef]
- Gunnell, A.; Webb, H.M.; Wood, C.D.; McClellan, M.J.; Wichaidit, B.; Kempkes, B.; Jenner, R.G.; Osborne, C.; Farrell, P.J.; West, M.J. RUNX super-enhancer control through the Notch pathway by Epstein-Barr virus transcription factors regulates B cell growth. Nucleic Acids Res. 2016, 44, 4636–4650. [Google Scholar] [CrossRef]
- Hosoi, H.; Niibori-Nambu, A.; Nah, G.S.S.; Bahirvani, A.G.; Mok, M.M.H.; Sanda, T.; Kumar, A.P.; Tenen, D.G.; Ito, Y.; Sonoki, T.; et al. Super-enhancers for RUNX3 are required for cell proliferation in EBV-infected B cell lines. Gene 2021, 774, 145421. [Google Scholar] [CrossRef]
- Frost, T.C.; Gewurz, B.E. Epigenetic crossroads of the Epstein-Barr virus B-cell relationship. Curr. Opin. Virol. 2018, 32, 15–23. [Google Scholar] [CrossRef]
- Hutchings, I.A.; Tierney, R.J.; Kelly, G.L.; Stylianou, J.; Rickinson, A.B.; Bell, A.I. Methylation Status of theEpstein-Barr Virus (EBV) BamHI W Latent Cycle Promoter and Promoter Activity: Analysis with Novel EBV-Positive Burkitt and LymphoblastoidCell Lines. J. Virol. 2006, 80, 10700–10711. [Google Scholar] [CrossRef]
- Li, H.; Minarovits, J. Host Cell-Dependent Expression of Latent Epstein–Barr Virus Genomes: Regulation by DNA Methylation. Adv. Cancer Res. 2003, 89, 133–156. [Google Scholar] [CrossRef]
- Park, J.-H.; Jeon, J.-P.; Shim, S.-M.; Nam, H.-Y.; Kim, J.-W.; Han, B.-G.; Lee, S. Wp specific methylation of highly proliferated LCLs. Biochem. Biophys. Res. Commun. 2007, 358, 513–520. [Google Scholar] [CrossRef]
- Robertson, K.D.; Manns, A.; Swinnen, L.J.; Zong, J.C.; Gulley, M.L.; Ambinder, R.F. CpG methylation of the major Epstein-Barr virus latency promoter in Burkitt’s lymphoma and Hodgkin’s disease. Blood 1996, 88, 3129–3136. [Google Scholar] [CrossRef]
- Schaefer, B.C.; Strominger, J.L.; Speck, S.H. Host-cell-determined methylation of specific Epstein-Barr virus promoters regulates the choice between distinct viral latency programs. Mol. Cell. Biol. 1997, 17, 364–377. [Google Scholar] [CrossRef][Green Version]
- Tao, Q.; Robertson, K.D.; Manns, A.; Hildesheim, A.; Ambinder, R.F. The Epstein-Barr virus major latent promoter Qp is constitutively active, hypomethylated, and methylation sensitive. J. Virol. 1998, 72, 7075–7083. [Google Scholar] [CrossRef]
- Tierney, R.J.; Kirby, H.E.; Nagra, J.K.; Desmond, J.; Bell, A.I.; Rickinson, A.B. Methylation of Transcription Factor Binding Sites in the Epstein-Barr Virus Latent Cycle Promoter Wp Coincides with Promoter Down-Regulation during Virus-Induced B-Cell Transformation. J. Virol. 2000, 74, 10468–10479. [Google Scholar] [CrossRef][Green Version]
- Falk, K.I.; Szekely, L.; Aleman, A.; Ernberg, I. Specific methylation patterns in two control regions of Epstein-Barr virus latency: The LMP-1-coding upstream regulatory region and an origin of DNA replication (oriP). J. Virol. 1998, 72, 2969–2974. [Google Scholar] [CrossRef]
- Li, J.; Liu, X.; Liu, M.; Che, K.; Luo, B. Methylation and expression of Epstein-Barr virus latent membrane protein 1, 2A and 2B in EBV-associated gastric carcinomas and cell lines. Dig. Liver Dis. 2016, 48, 673–680. [Google Scholar] [CrossRef]
- Lu, F.; Wiedmer, A.; Martin, K.A.; Wickramasinghe, P.J.M.S.; Kossenkov, A.V.; Lieberman, P.M. Coordinate Regulation of TET2 and EBNA2 Controls the DNA Methylation State of Latent Epstein-Barr Virus. J. Virol. 2017, 91, e00804-17. [Google Scholar] [CrossRef]
- Wille, C.K.; Li, Y.; Rui, L.; Johannsen, E.C.; Kenney, S.C. Restricted TET2 Expression in Germinal Center Type B Cells Promotes Stringent Epstein-Barr Virus Latency. J. Virol. 2017, 91, e01987-16. [Google Scholar] [CrossRef]
- Guo, R.; Zhang, Y.; Teng, M.; Jiang, C.; Schineller, M.; Zhao, B.; Doench, J.G.; O’Reilly, R.J.; Cesarman, E.; Giulino-Roth, L.; et al. DNA methylation enzymes and PRC1 restrict B-cell Epstein–Barr virus oncoprotein expression. Nat. Microbiol. 2020, 5, 1051–1063. [Google Scholar] [CrossRef] [PubMed]
- Herold, M.; Bartkuhn, M.; Renkawitz, R. CTCF: Insights into insulator function during development. Development 2012, 139, 1045–1057. [Google Scholar] [CrossRef] [PubMed]
- Hnisz, D.; Day, D.S.; Young, R.A. Insulated Neighborhoods: Structural and Functional Units of Mammalian Gene Control. Cell 2016, 167, 1188–1200. [Google Scholar] [CrossRef]
- Tempera, I.; Wiedmer, A.; Dheekollu, J.; Lieberman, P.M. CTCF Prevents the Epigenetic Drift of EBV Latency Promoter Qp. PLoS Pathog. 2010, 6, e1001048. [Google Scholar] [CrossRef]
- Arvey, A.; Tempera, I.; Tsai, K.; Chen, H.-S.; Tikhmyanova, N.; Klichinsky, M.; Leslie, C.; Lieberman, P.M. An Atlas of the Epstein-Barr Virus Transcriptome and Epigenome Reveals Host-Virus Regulatory Interactions. Cell Host Microbe 2012, 12, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Tempera, I.; Klichinsky, M.; Lieberman, P.M. EBV Latency Types Adopt Alternative Chromatin Conformations. PLoS Pathog. 2011, 7, e1002180. [Google Scholar] [CrossRef]
- Tempera, I.; Lieberman, P.M. Epigenetic regulation of EBV persistence and oncogenesis. Semin. Cancer Biol. 2014, 26, 22–29. [Google Scholar] [CrossRef]
- Arvey, A.; Tempera, I.; Lieberman, P.M. Interpreting the Epstein-Barr Virus (EBV) Epigenome Using High-Throughput Data. Viruses 2013, 5, 1042–1054. [Google Scholar] [CrossRef]
- Day, L.; Chau, C.M.; Nebozhyn, M.; Rennekamp, A.J.; Showe, M.; Lieberman, P.M. Chromatin Profiling of Epstein-Barr Virus Latency Control Region. J. Virol. 2007, 81, 6389–6401. [Google Scholar] [CrossRef] [PubMed]
- Imai, K.; Kamio, N.; Cueno, M.E.; Saito, Y.; Inoue, H.; Saito, I.; Ochiai, K. Role of the histone H3 lysine 9 methyltransferase Suv39 h1 in maintaining Epsteinn-Barr virus latency in B95-8 cells. FEBS J. 2014, 281, 2148–2158. [Google Scholar] [CrossRef]
- Murata, T.; Kondo, Y.; Sugimoto, A.; Kawashima, D.; Saito, S.; Isomura, H.; Kanda, T.; Tsurumi, T. Epigenetic Histone Modification of Epstein-Barr Virus BZLF1 Promoter during Latency and Reactivation in Raji Cells. J. Virol. 2012, 86, 4752–4761. [Google Scholar] [CrossRef]
- Ramasubramanyan, S.; Osborn, K.; Flower, K.; Sinclair, A.J. Dynamic Chromatin Environment of Key Lytic Cycle Regulatory Regions of the Epstein-Barr Virus Genome. J. Virol. 2012, 86, 1809–1819. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Li, F.; Wan, M.; Zhang, B.; Peng, Y.; Zhou, Y.; Pi, C.; Xu, X.; Ye, L.; Zhou, X.; Zheng, L. Bivalent Histone Modifications and Development. Curr. Stem Cell Res. Ther. 2018, 13, 83–90. [Google Scholar] [CrossRef]
- Djavadian, R.; Chiu, Y.-F.; Johannsen, E. An Epstein-Barr Virus-Encoded Protein Complex Requires an Origin of Lytic Replication In Cis to Mediate Late Gene Transcription. PLoS Pathog. 2016, 12, e1005718. [Google Scholar] [CrossRef]
- Li, H.; Liu, S.; Hu, J.; Luo, X.; Li, N.; Bode, A.M.; Cao, Y. Epstein-Barr virus lytic reactivation regulation and its pathogenic role in carcinogenesis. Int. J. Biol. Sci. 2016, 12, 1309–1318. [Google Scholar] [CrossRef]
- Münz, C. Latency and lytic replication in Epstein–Barr virus-associated oncogenesis. Nat. Rev. Microbiol. 2019, 17, 691–700. [Google Scholar] [CrossRef]
- Munz, C. The Role of Lytic Infection for Lymphomagenesis of Human gamma-Herpesviruses. Front. Cell. Infect. Microbiol. 2021, 11, 605258. [Google Scholar] [CrossRef]
- Murata, T.; Okuno, Y.; Sato, Y.; Watanabe, T.; Kimura, H. Oncogenesis of CAEBV revealed: Intragenic deletions in the viral genome and leaky expression of lytic genes. Rev. Med. Virol. 2020, 30, e2095. [Google Scholar] [CrossRef]
- Wu, C.-C.; Fang, C.-Y.; Huang, S.-Y.; Chiu, S.-H.; Lee, C.-H.; Chen, J.-Y. Perspective: Contribution of Epstein–Barr virus (EBV) Reactivation to the Carcinogenicity of Nasopharyngeal Cancer Cells. Cancers 2018, 10, 120. [Google Scholar] [CrossRef]
- Katsumura, K.R.; Maruo, S.; Takada, K. EBV lytic infection enhances transformation of B-lymphocytes infected with EBV in the presence of T-lymphocytes. J. Med. Virol. 2012, 84, 504–510. [Google Scholar] [CrossRef]
- Kimura, H.; Okuno, Y.; Sato, Y.; Watanabe, T.; Murata, T. Deletion of Viral microRNAs in the Oncogenesis of Epstein–Barr Virus-Associated Lymphoma. Front. Microbiol. 2021, 12, 667968. [Google Scholar] [CrossRef]
- Okuno, Y.; Murata, T.; Sato, Y.; Muramatsu, H.; Ito, Y.; Watanabe, T.; Okuno, T.; Murakami, N.; Yoshida, K.; Sawada, A.; et al. Defective Epstein–Barr virus in chronic active infection and haematological malignancy. Nat. Microbiol. 2019, 4, 404–413. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.-D.; Hegde, S.; Young, K.H.; Sullivan, R.; Rajesh, D.; Zhou, Y.; Jankowska-Gan, E.; Burlingham, W.J.; Sun, X.; Gulley, M.L.; et al. A New Model of Epstein-Barr Virus Infection Reveals an Important Role for Early Lytic Viral Protein Expression in the Development of Lymphomas. J. Virol. 2011, 85, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.-D.; Yu, X.; Mertz, J.E.; Gumperz, J.E.; Reinheim, E.; Zhou, Y.; Tang, W.; Burlingham, W.J.; Gulley, M.L.; Kenney, S.C. An Epstein-Barr Virus (EBV) Mutant with Enhanced BZLF1 Expression Causes Lymphomas with Abortive Lytic EBV Infection in a Humanized Mouse Model. J. Virol. 2012, 86, 7976–7987. [Google Scholar] [CrossRef] [PubMed]
- Martini, M.; Capello, D.; Serraino, D.; Navarra, A.; Pierconti, F.; Cenci, T.; Gaidano, G.; Larocca, L.M. Characterization of variants in the promoter of EBV gene BZLF1 in normal donors, HIV-positive patients and in AIDS-related lymphomas. J. Infect. 2007, 54, 298–306. [Google Scholar] [CrossRef] [PubMed]
- Tong, J.H.M.; Lo, K.W.; Au, F.W.L.; Huang, D.P.; To, K.-F. Re: Discrete Alterations in the BZLF1 Promoter in Tumor and Non-Tumor-Associated Epstein-Barr Virus. J. Natl. Cancer Inst. 2003, 95, 1008–1009. [Google Scholar] [CrossRef]
- Bristol, J.A.; Djavadian, R.; Albright, E.R.; Coleman, C.B.; Ohashi, M.; Hayes, M.; Romero-Masters, J.C.; Barlow, E.A.; Farrell, P.J.; Rochford, R.; et al. A cancer-associated Epstein-Barr virus BZLF1 promoter variant enhances lytic infection. PLoS Pathog. 2018, 14, e1007179. [Google Scholar] [CrossRef]
- Palser, A.L.; Grayson, N.E.; White, R.E.; Corton, C.; Correia, S.; Ba Abdullah, M.M.; Watson, S.J.; Cotten, M.; Arrand, J.R.; Murray, P.G.; et al. Genome Diversity of Epstein-Barr Virus from Multiple Tumor Types and Normal Infection. J. Virol. 2015, 89, 5222–5237. [Google Scholar] [CrossRef]
- Venturini, C.; Houldcroft, C.J.; Lazareva, A.; Wegner, F.; Morfopoulou, S.; Amrolia, P.J.; Golwala, Z.; Rao, A.; Marks, S.D.; Simmonds, J.; et al. Epstein–Barr virus (EBV) deletions as biomarkers of response to treatment of chronic active EBV. Br. J. Haematol. 2021, 195, 249–255. [Google Scholar] [CrossRef]
- Lin, X.; Tsai, M.-H.; Shumilov, A.; Poirey, R.; Bannert, H.; Middeldorp, J.; Feederle, R.; Delecluse, H.-J. The Epstein-Barr Virus BART miRNA Cluster of the M81 Strain Modulates Multiple Functions in Primary B Cells. PLoS Pathog. 2015, 11, e1005344. [Google Scholar] [CrossRef] [PubMed]
- Stuart, A.D.; Stewart, J.P.; Arrand, J.R.; Mackett, M. The Epstein-Barr virus encoded cytokine viral interleukin-10 enhances transformation of human B lymphocytes. Oncogene 1995, 11, 1711–1720. [Google Scholar] [PubMed]
- Murata, T.; Iwata, S.; Alam Siddiquey, M.N.; Kanazawa, T.; Goshima, F.; Kawashima, D.; Kimura, H.; Tsurumi, T. Heat Shock Protein 90 Inhibitors Repress Latent Membrane Protein 1 (LMP1) Expression and Proliferation of Epstein-Barr Virus-Positive Natural Killer Cell Lymphoma. PLoS ONE 2013, 8, e63566. [Google Scholar] [CrossRef]
- Suzuki, M.; Takeda, T.; Nakagawa, H.; Iwata, S.; Watanabe, T.; Siddiquey, M.N.; Goshima, F.; Murata, T.; Kawada, J.I.; Ito, Y.; et al. The heat shock protein 90 inhibitor BIIB021 suppresses the growth of T and natural killer cell lymphomas. Front. Microbiol. 2015, 6, 280. [Google Scholar] [CrossRef] [PubMed]
- Iwata, S.; Saito, T.; Ito, Y.; Kamakura, M.; Gotoh, K.; Kawada, J.-I.; Nishiyama, Y.; Kimura, H. Antitumor activities of valproic acid on Epstein-Barr virus-associated T and natural killer lymphoma cells. Cancer Sci. 2012, 103, 375–381. [Google Scholar] [CrossRef]
- Siddiquey, M.N.; Nakagawa, H.; Iwata, S.; Kanazawa, T.; Suzuki, M.; Imadome, K.; Fujiwara, S.; Goshima, F.; Murata, T.; Kimura, H. Anti-tumor effects of suberoylanilide hydroxamic acid on Epstein–Barr virus-associated T cell and natural killer cell lymphoma. Cancer Sci. 2014, 105, 713–722. [Google Scholar] [CrossRef]
- Nishikawa, J.; Iizasa, H.; Yoshiyama, H.; Nakamura, M.; Saito, M.; Sasaki, S.; Shimokuri, K.; Yanagihara, M.; Sakai, K.; Suehiro, Y.; et al. The Role of Epigenetic Regulation in Epstein-Barr Virus-Associated Gastric Cancer. Int. J. Mol. Sci. 2017, 18, 1606. [Google Scholar] [CrossRef]
- Chan, A.T.; Tao, Q.; Robertson, K.D.; Flinn, I.W.; Mann, R.B.; Klencke, B.; Kwan, W.H.; Leung, T.W.-T.; Johnson, P.J.; Ambinder, R.F. Azacitidine Induces Demethylation of the Epstein-Barr Virus Genome in Tumors. J. Clin. Oncol. 2004, 22, 1373–1381. [Google Scholar] [CrossRef] [PubMed]
- Dalton, T.; Doubrovina, E.; Pankov, D.; Reynolds, I.R.C.; Scholze, H.; Selvakumar, A.; Vizconde, T.; Savalia, B.; Dyomin, V.; Weigel, C.; et al. Epigenetic reprogramming sensitizes immunologically silent EBV+ lymphomas to virus-directed immunotherapy. Blood 2020, 135, 1870–1881. [Google Scholar] [CrossRef] [PubMed]
- Yiu, S.P.T.; Dorothea, M.; Hui, K.F.; Chiang, A.K.S. Lytic Induction Therapy against Epstein-Barr Virus-Associated Malignancies: Past, Present, and Future. Cancers 2020, 12, 2142. [Google Scholar] [CrossRef]
- Ghosh, S.K.; Perrine, S.P.; Williams, R.M.; Faller, D.V. Histone deacetylase inhibitors are potent inducers of gene expression in latent EBV and sensitize lymphoma cells to nucleoside antiviral agents. Blood 2012, 119, 1008–1017. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sub-Family | Site of Latency | Viral Gene Expressed in Latency | Trigger of Reactivation | Viral Lytic Swithch Gene | Transcriptional Activator of Reactivation | Transcriptional Repressor of Reactivation | |
|---|---|---|---|---|---|---|---|
| HSV-1 | a | neuron cells in trigeminal ganglia | LATs | stressors, immune suppression | ICP0, ICP4 | HCF1, SP1, GR, KLF15, ROS | NGF |
| VZV | neuron cells in dorsal root ganglia | ORF63, VLT | stressors, immune suppression | IE62 | HCF1, SP1, YY1, PI3K/AKT, JNK | ||
| HCMV | b | myeloid lineages, hematopoietic progenitor cells, monocytes | UL138, UL81-82ast(LUNA), US28, UL144, UL111A(vIL10) | stressors, immune suppression, differentiation, growth factor | IE86(UL122), IE72(UL123) | PU.1, SP1, C/EBP, ATF/CREB, AP-1, NFkB, PPARg, RAR/RXR | CUX1/CDP, GFI-1, YY1, CTCF |
| EBV | g | B cells, epithelial cells | EBNAs, LMPs, EBERs | stressors, immune suppression, differentiation, groth factor, hypoxia | BZLF1(Zta), BRLF1(Rta) | SP1, KLF4, MEF2, C/EBP, ATF/CREB, AP-1, XBP1s, SMAD, HIF, ROS, P53, PI3K/AKT, TORC2, YAP/TAZ, TET2, ATM, BLIMP1, TAF-I/NAP1, NFATc1, CASP1 | ZEB, NFkB, MYC, YY1, JDP2, NO, E2-2, JAK/STAT, SMUBP, OCT2, PAX5, BCL6, ARKL1, PARP1, CAF1/HIRA, KAP1 |
| KSHV | B cells, endothelial cells | LANA, vCYC, vFLIP, Kaposins | stressors, immune suppression, differentiation, groth factor, hypoxia | K-Rta (ORF50) | C/EBP, AP-1, EGR1, XBP1, NFAT, HIF, ROS | NFkB, MYC, KAP1, PI3K/AKT, IFNs, IRFs, HES1, FOXO1 |
| Patterns | EBERs | EBNA1 | LMP1/2 | EBNA2/3/LP |
|---|---|---|---|---|
| Latency III | + | + | + | + |
| Latency IIa | + | + | + | |
| Latency IIb | + | + | + | |
| Latency I | + | + | ||
| Latency 0 | + |
| EBV | Necessity # | HSV | HCMV | Function |
|---|---|---|---|---|
| BZLF1 * | essential | UL9 * | UL84 * | oriLyt-binding |
| BALF5 | essential | UL30 | UL54 | polymerase catalytic subunit (Pol) |
| BMRF1 | essential | UL42 | UL44 | processivity subunit of polymerase |
| BALF2 | essential | UL29 | UL57 | single-stranded DNA-binding protein (ssDNABP) |
| BBLF4 | essential | UL5 | UL105 | helicase |
| BSLF1 | essential | UL52 | UL70 | primase |
| BBLF2/3 | essential | UL8 | UL102 | primase-binding protein |
| BKRF3 | essential | UL29 | UL114 | uracil-DNA glycosylase, involved in DNA repair |
| BXLF1 | supportive | UL23 | – | thymidine kinase (TK) |
| BORF2 | supportive | UL39 | UL45 | ribonucleotide reductase (RR) large subunit |
| BaRF1 | supportive | UL40 | – | ribonucleotide reductase (RR) small subunit |
| BLLF3 | supportive | UL50 | UL72 | deoxyuridine triphosphate nuclotidohydrolase (dUTPase) |
| EBV | HCMV | Presumed Function and/or Nature |
|---|---|---|
| BcRF1 | UL87 | TATT-binding protein, associates with RNAPII |
| BDLF3.5 | UL91 | |
| BDLF4 | UL92 | stabilized by phosphorylation |
| BFRF2 | UL49 | DNA-binding protein, having potential zinc-finger domain |
| BGLF3 | UL95 | hub of vPIC, phosphorylation increased association with BFRF2 and BVLF1 |
| BVLF1 | UL79 | elongation factor (in HCMV) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murata, T.; Sugimoto, A.; Inagaki, T.; Yanagi, Y.; Watanabe, T.; Sato, Y.; Kimura, H. Molecular Basis of Epstein–Barr Virus Latency Establishment and Lytic Reactivation. Viruses 2021, 13, 2344. https://doi.org/10.3390/v13122344
Murata T, Sugimoto A, Inagaki T, Yanagi Y, Watanabe T, Sato Y, Kimura H. Molecular Basis of Epstein–Barr Virus Latency Establishment and Lytic Reactivation. Viruses. 2021; 13(12):2344. https://doi.org/10.3390/v13122344
Chicago/Turabian StyleMurata, Takayuki, Atsuko Sugimoto, Tomoki Inagaki, Yusuke Yanagi, Takahiro Watanabe, Yoshitaka Sato, and Hiroshi Kimura. 2021. "Molecular Basis of Epstein–Barr Virus Latency Establishment and Lytic Reactivation" Viruses 13, no. 12: 2344. https://doi.org/10.3390/v13122344
APA StyleMurata, T., Sugimoto, A., Inagaki, T., Yanagi, Y., Watanabe, T., Sato, Y., & Kimura, H. (2021). Molecular Basis of Epstein–Barr Virus Latency Establishment and Lytic Reactivation. Viruses, 13(12), 2344. https://doi.org/10.3390/v13122344