
Potential Effects of Human Papillomavirus Type Substitution, Superinfection Exclusion and Latency on the Efficacy of the Current L1 Prophylactic Vaccines


Abstract
1. Introduction
2. HPV Types and Associated Pathologies
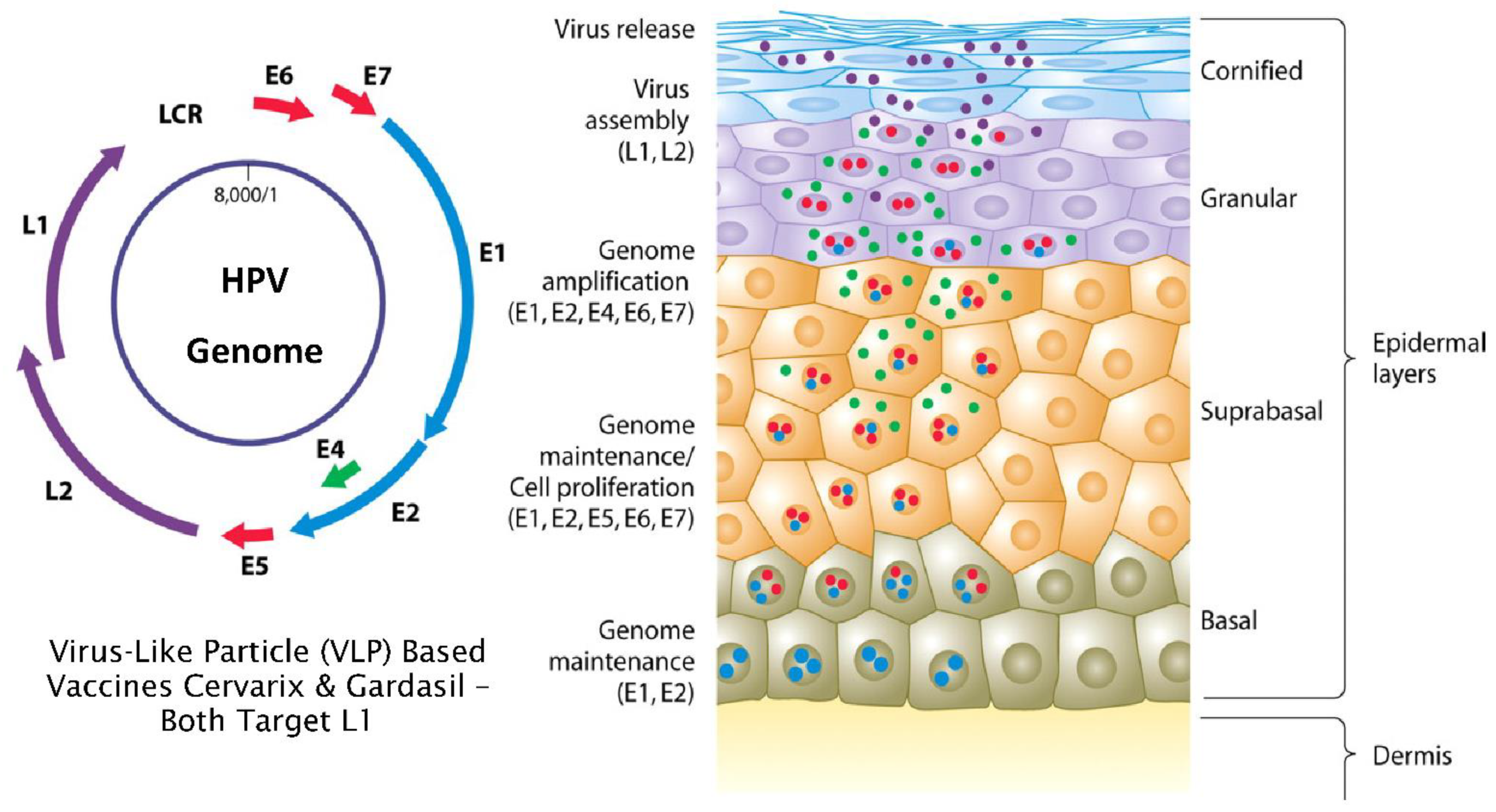
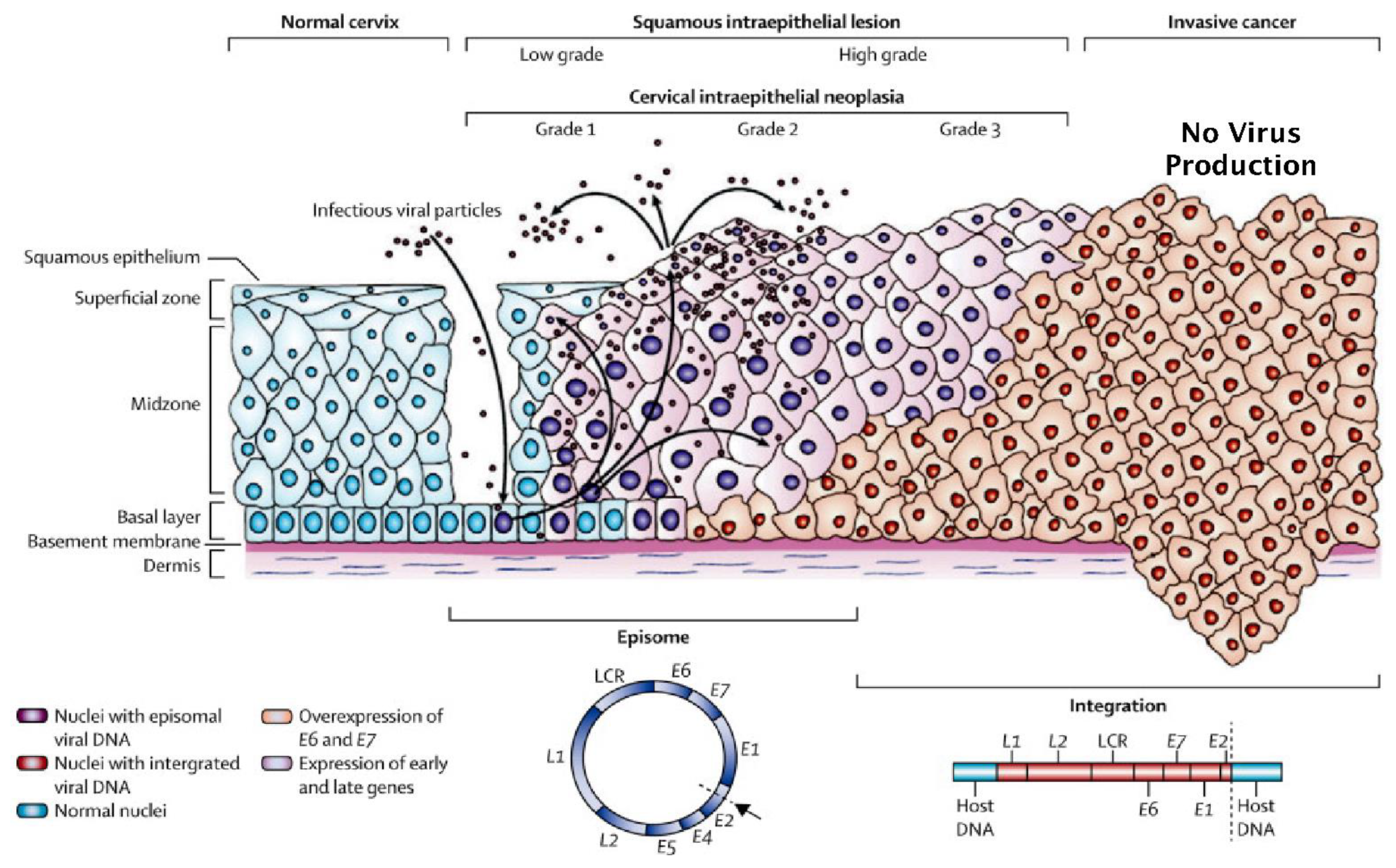
3. Normal versus Truncated HPV Life Cycles and the Development of Neoplasia
4. Mode of Action of L1 Targeted Prophylactic Vaccines
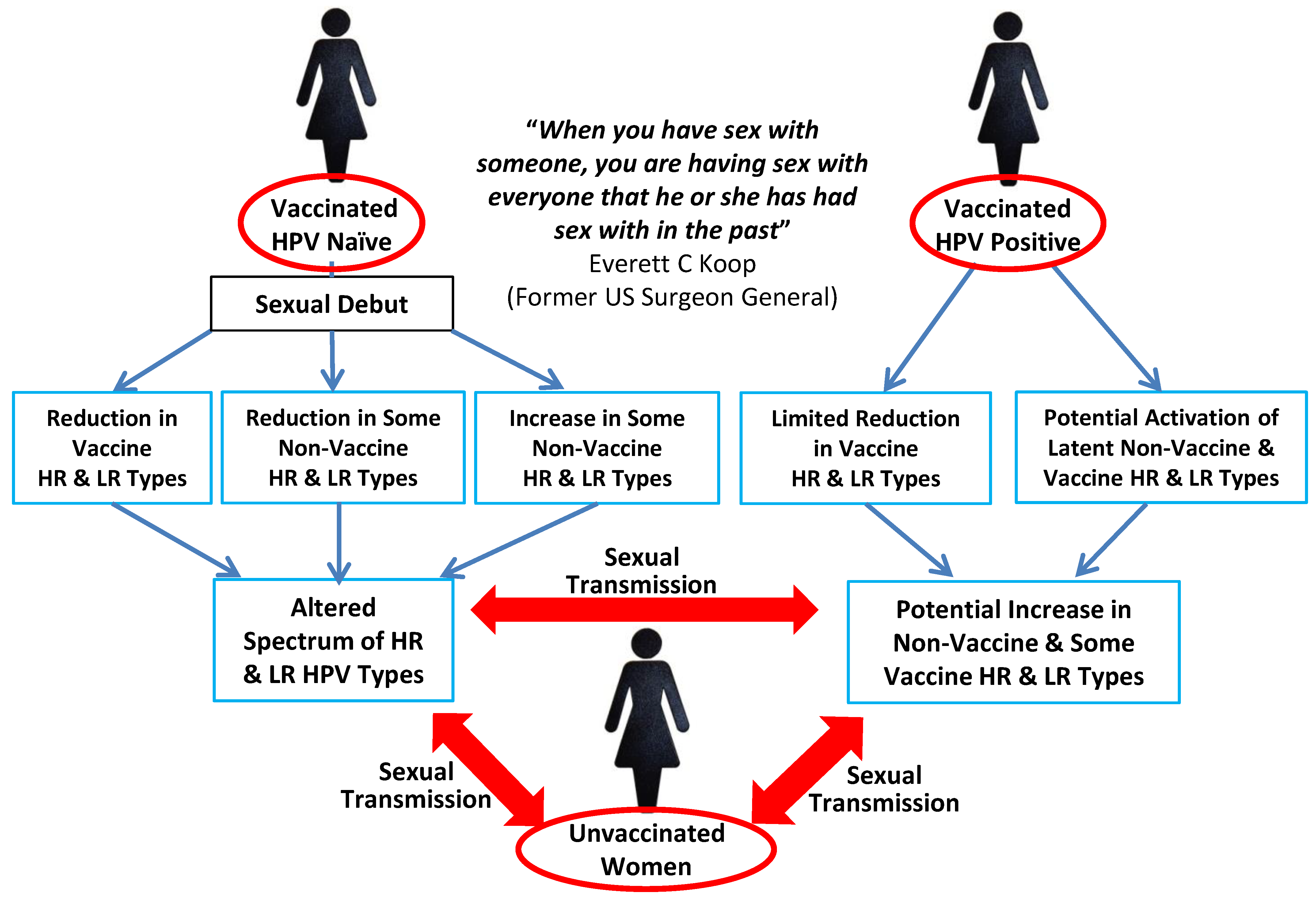
5. The Influence of HPV Superinfection, Superinfection Exclusion and Latency
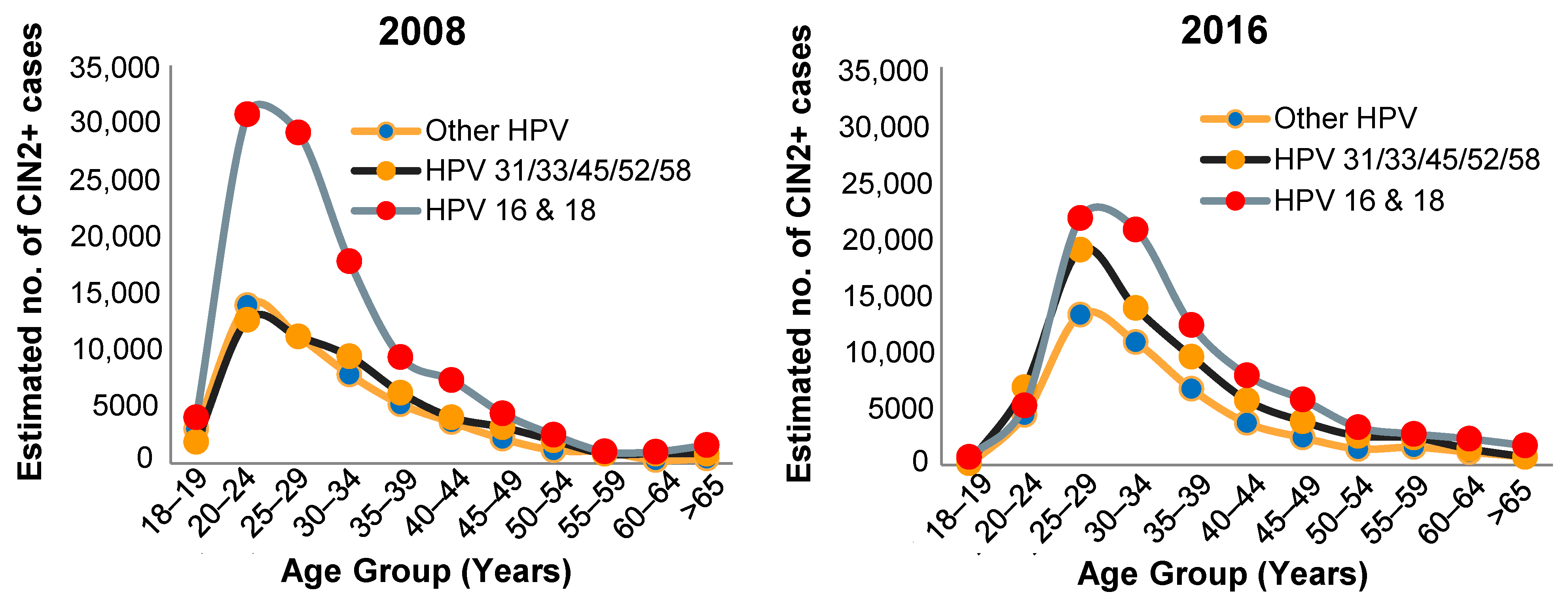
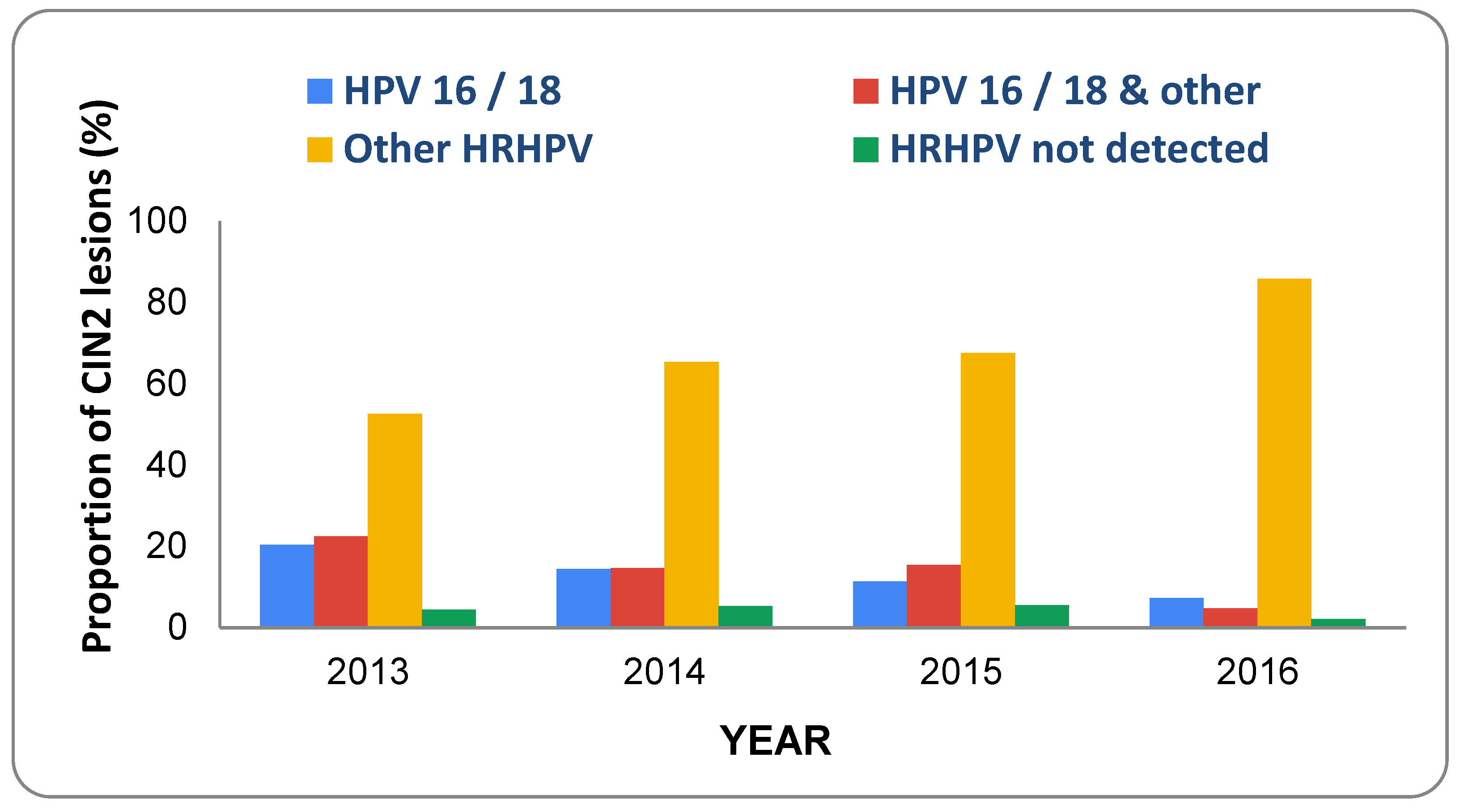
6. Prevalence of HPV Types and CIN in Vaccinated Populations
- Extent of vaccine efficacy in HPV-positive women.
- Extent of HPV cross-type protection.
- Extent of HPV type replacement and the potential role played by latent infections.
- Vaccinate prior to onset of sexual activity and begin assessment of end points at age of usual cervical screening once sexually active.
- Make all clinical study reports, including anonymised individual patient data, publicly available.
- Separate trials to assess benefits in women already exposed to HPV without restrictions based on risk factors.
- Analyse data by country and study site.
- Ensure the testing interval is in line with usual cervical screening protocols.
- Continue follow up for a minimum of 20 years from time of sexual debut.
- Power trials for primary composite outcomes CIN3/AIS/cervical cancer due to oncogenic HPV types.
- Define secondary outcome of persistent HPV16/18 infection at a minimum of 12 months.
- Use standardised methods for HPV testing.
- Undertake a saline placebo-controlled efficacy trial of Gardasil 9 in previously unvaccinated populations as it is difficult to draw conclusions on efficacy and risk of harms based on comparing Gardasil 9 to Gardasil.
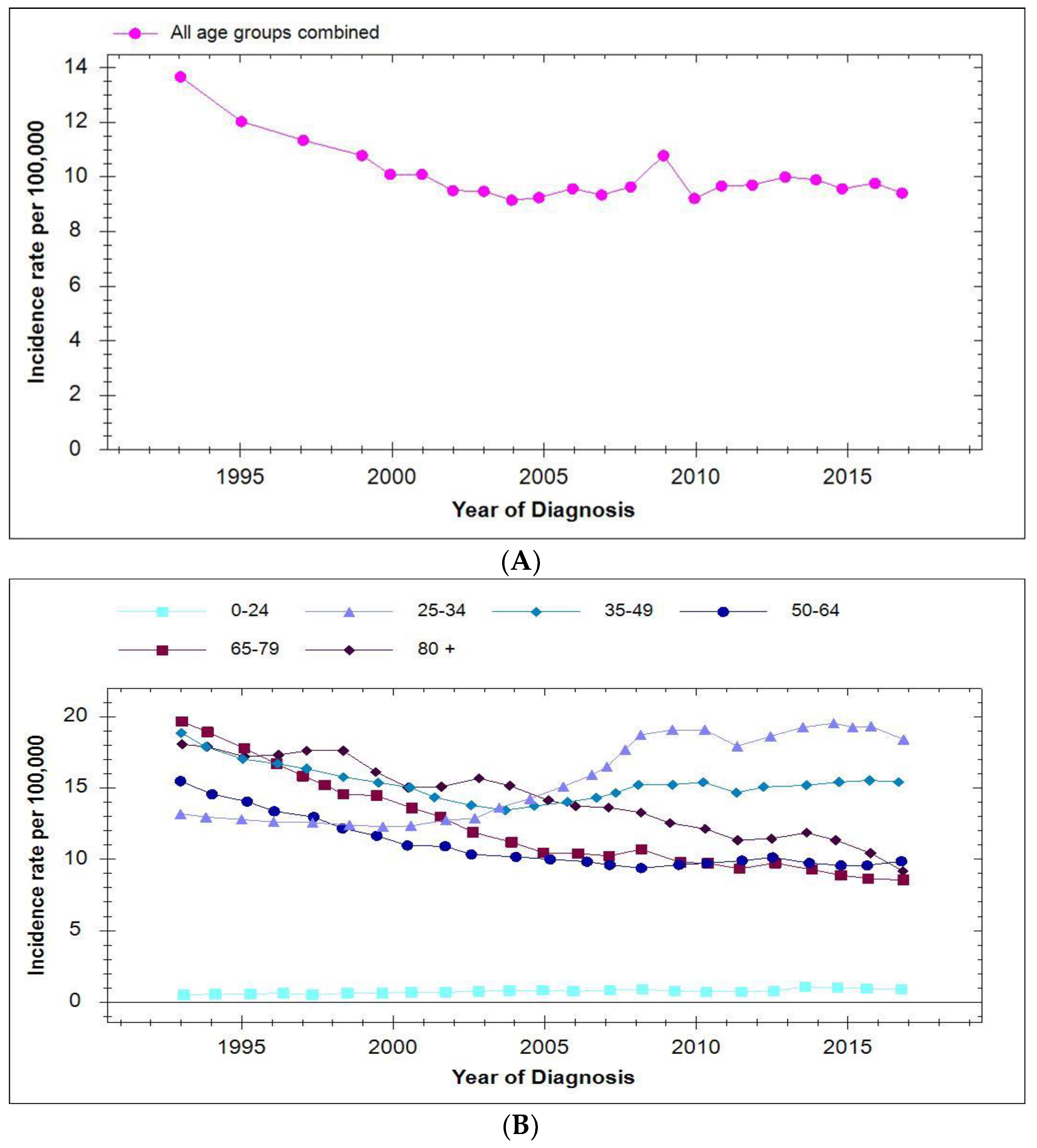
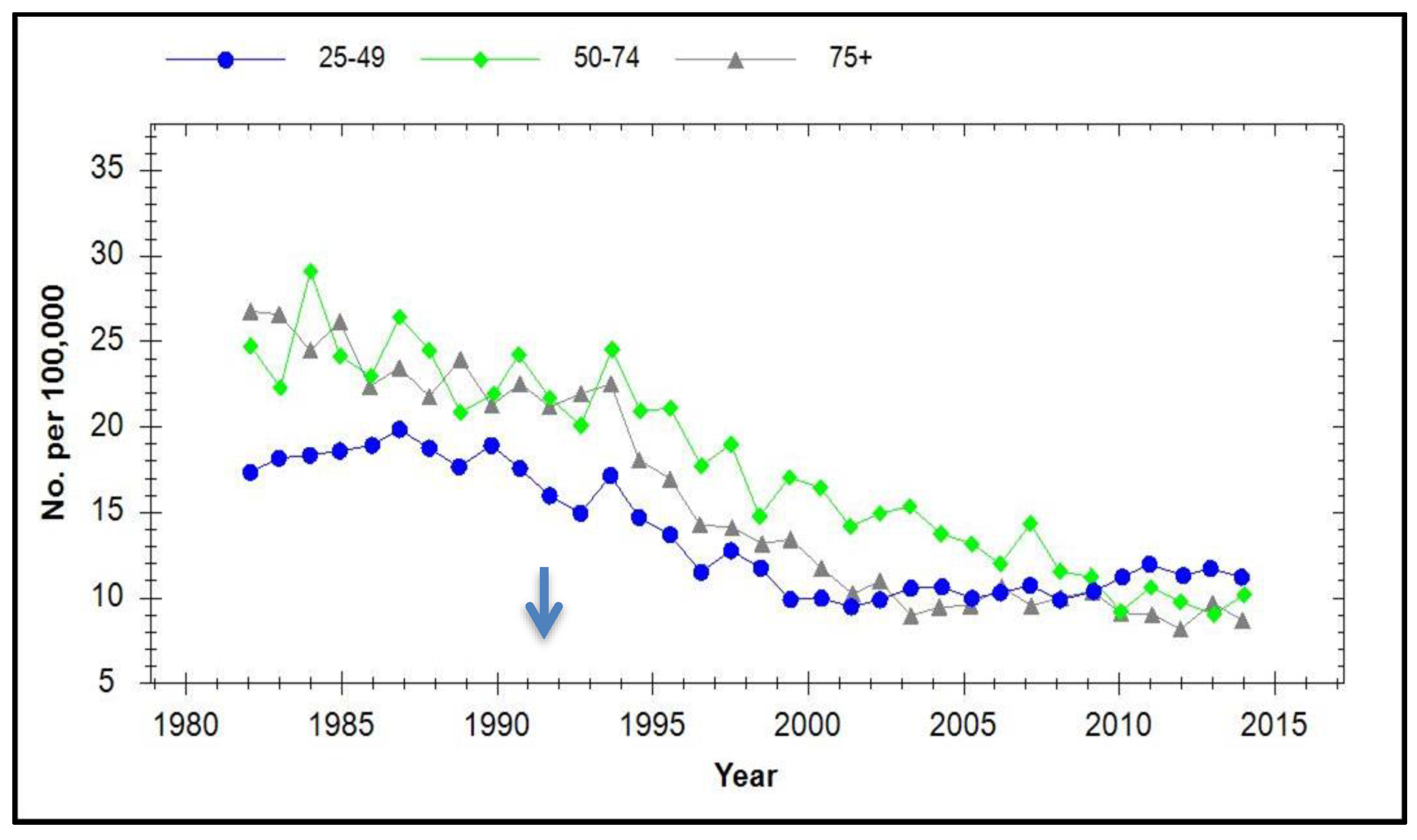
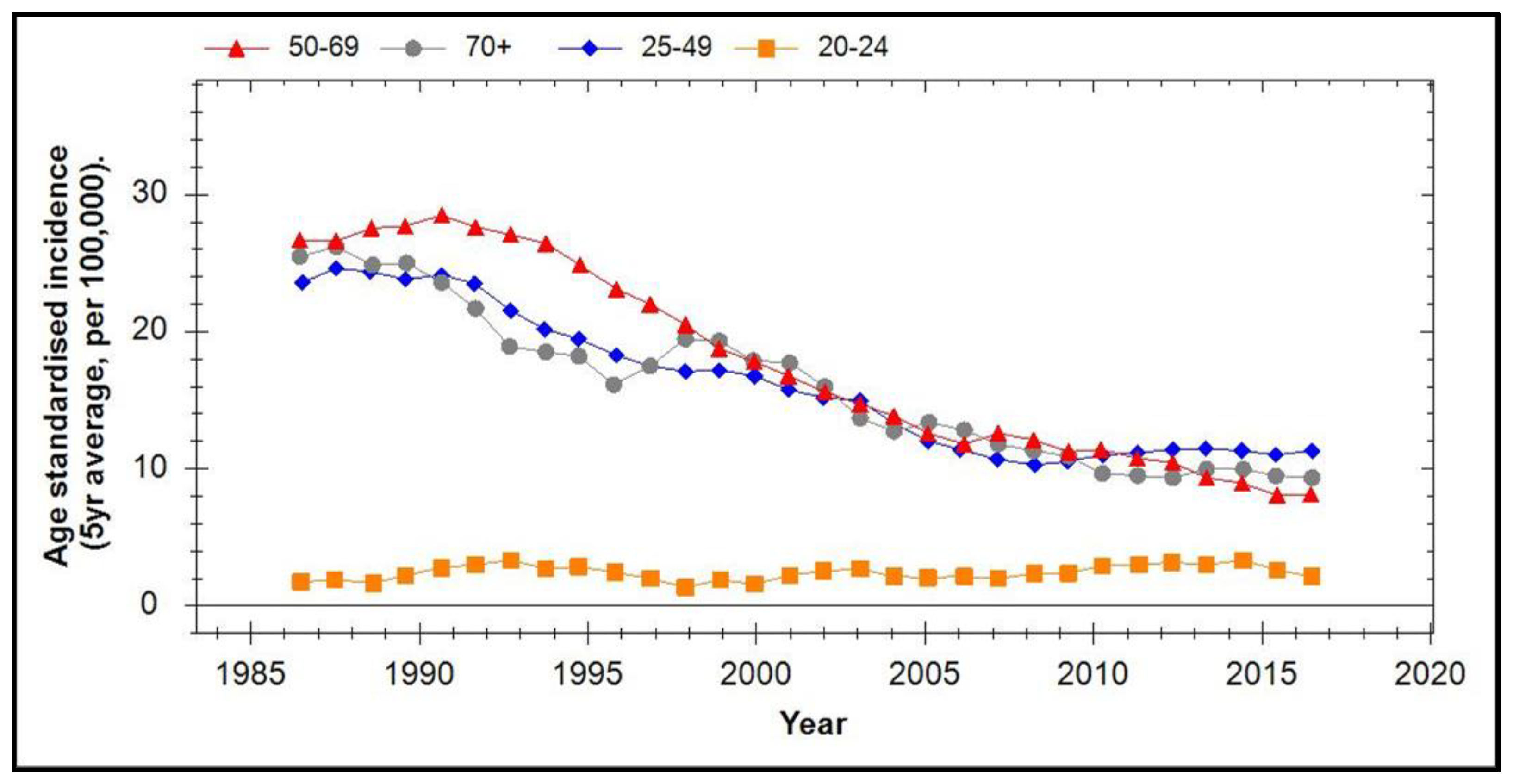
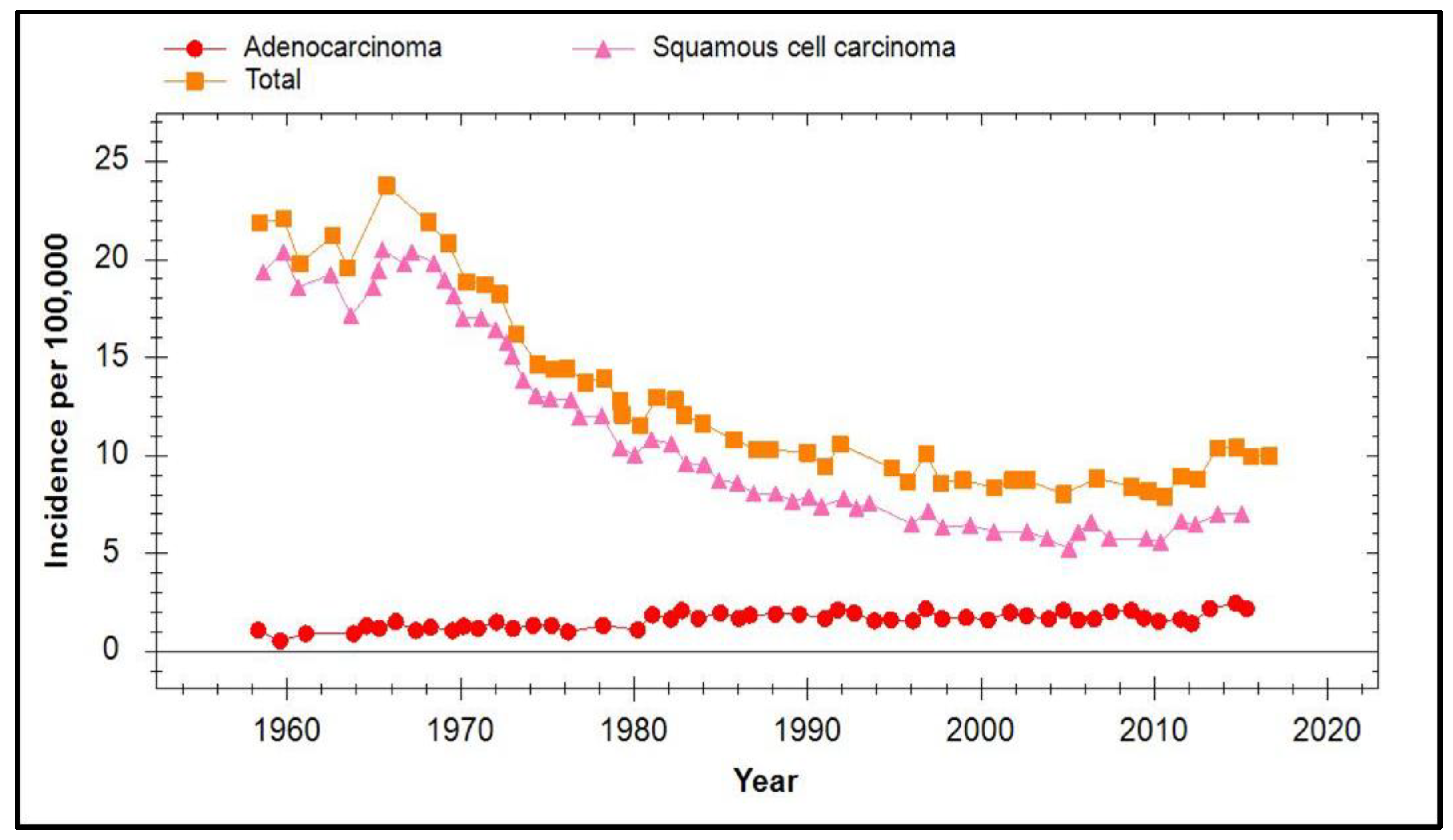
7. Incidence of Cervical Cancer in Vaccinated Populations
8. Discussion and Conclusions
- Use of high-sensitivity methods of HPV analysis in order to detect latent infections.
- Determination of the impact of vaccination over time on the prevalence of all 51 HPV types known to infect genital mucosa in all age groups and combine this with vaccination status, age at vaccination, vaccine type and sexual history.
- Use of this to discriminate between direct and indirect (herd) effects of vaccination on the incidence of different HPV types in unvaccinated women.
- Carry out these analyses on normal, CIN1, 2 and 3 and cervical cancers.
- Do not assume that unvaccinated women residing in a vaccinated population will experience only beneficial herd effects since increased transmission of non-vaccine-covered high-risk HPV types combined with decreased transmission of low-risk types has been shown to occur [74].
- Conclusions on vaccine efficacy should not be based on comparing vaccinated and unvaccinated women over time at the same geographical location since herd-effect transmission may confound this.
- Ideally, the incidence and disease association of different HPV types should be evaluated in any population before and after implementing vaccination.
- Changes in sexual practice.
- Changes in screening practice.
- Problems with assay sensitivity.
- Influx of migrant communities.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colafrancesco, S.; Perricone, C.; Tomljenovic, L.; Shoenfeld, Y. Human Papilloma Virus Vaccine and Primary Ovarian Failure: Another Facet of the Autoimmune/Inflammatory Syndrome Induced by Adjuvants. Am. J. Reprod. Immunol. 2013, 70, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Ji, H.-H.; Tang, X.-W.; Pan, L.-Y.; Chen, X.; Jia, Y.-T. Human papillomavirus vaccine-associated premature ovarian insufficiency and related adverse events: Data mining of Vaccine Adverse Event Reporting System. Sci. Rep. 2020, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A.B.; Fernandes, E.G.; Miyaji, K.T.; Arantes, B.A.; Valente, M.G.; Sato, H.K.; Sartori, A.M.C. Adverse events following Quadrivalent HPV vaccination reported in Sao Paulo State, Brazil, in the first three years after introducing the vaccine for routine immunization (March 2014 to December 2016). Revista do Instituto de Medicina Tropical de São Paulo 2019, 61, 43. [Google Scholar] [CrossRef] [PubMed]
- Phillips, A.; Hickie, M.; Totterdell, J.; Brotherton, J.M.; Dey, A.; Hill, R.; Snelling, T.; Macartney, K. Adverse events following HPV vaccination: 11 years of surveillance in Australia. Vaccine 2020, 38, 6038–6046. [Google Scholar] [CrossRef]
- Geier, D.A.; Geier, M.R. Quadrivalent human papillomavirus vaccine and autoimmune adverse events: A case-control as-sessment of the vaccine adverse event reporting system (VAERS) database. Immunol. Res. 2017, 65, 46–54. [Google Scholar] [CrossRef]
- Burd, E.M. Human Papillomavirus and Cervical Cancer. Clin. Microbiol. Rev. 2003, 16, 1–17. [Google Scholar] [CrossRef]
- Steinbach, A.; Riemer, A.B. Immune evasion mechanisms of human papillomavirus: An update. Int. J. Cancer 2018, 142, 224–229. [Google Scholar] [CrossRef]
- Brancaccio, R.N.; Robitaille, A.; Dutta, S.; Rollison, D.E.; Tommasino, M.; Gheit, T. Isolation of a Novel Beta-2 Human Pap-illomavirus from Skin. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef]
- Schmitt, M.; Depuydt, C.; Benoy, I.; Bogers, J.; Antoine, J.; Arbyn, M.; Pawlita, M.; on behalf of the VALGENT Study Group. Prevalence and viral load of 51 genital human papillomavirus types and three subtypes. Int. J. Cancer 2012, 132, 2395–2403. [Google Scholar] [CrossRef]
- Bonnez, W. Human Papillomavirus. In Vaccines for Biodefense and Emerging and Neglected Diseases; Alan, D.T., Barrett, L.R.S., Eds.; Academic Press: Cambridge, MA, USA, 2009; pp. 469–496. [Google Scholar]
- Moscicki, A.B.; Palefsky, J.M. Human papillomavirus in men: An update. J. Low. Genit. Tract Dis. 2011, 15, 231–234. [Google Scholar] [CrossRef]
- Preti, M.; Rotondo, J.C.; Holzinger, D.; Micheletti, L.; Gallio, N.; McKay-Chopin, S.; Carreira, C.; Privitera, S.S.; Watanabe, R.; Ridder, R.; et al. Role of human papillomavirus infection in the etiology of vulvar cancer in Italian women. Infect. Agents Cancer 2020, 15, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Bzhalava, Z.; Mühr, L.S.A.; Dillner, J. Transcription of human papillomavirus oncogenes in head and neck squamous cell carcinomas. Vaccine 2020, 38, 4066–4070. [Google Scholar] [CrossRef] [PubMed]
- Del Prete, R.; Luigi, R.; Magrone, R.; Addati, G.; Abbasciano, A.; Di Carlo, D.; Miragliotta, G. Epidemiological evaluation of human papillomavirus genotypes and their associations in multiple infections. Epidemiol. Infect. 2019, 147, e132. [Google Scholar] [CrossRef] [PubMed]
- Stanley, M.A. Epithelial Cell Responses to Infection with Human Papillomavirus. Clin. Microbiol. Rev. 2012, 25, 215–222. [Google Scholar] [CrossRef]
- Lazarczyk, M.; Cassonnet, P.; Pons, C.; Jacob, Y.; Favre, M. The EVER Proteins as a Natural Barrier against Papillomaviruses: A New Insight into the Pathogenesis of Human Papillomavirus Infections. Microbiol. Mol. Biol. Rev. 2009, 73, 348–370. [Google Scholar] [CrossRef]
- Woodman, C.B.; Collins, S.I.; Young, L.S. The natural history of cervical HPV infection: Unresolved issues. Nat. Rev. Cancer 2007, 7, 11–22. [Google Scholar] [CrossRef]
- Ho, C.M.; Lee, B.H.; Chang, S.F.; Chien, T.Y.; Huang, S.H.; Yan, C.C.; Cheng, W.F. Integration of human papilloma-virus correlates with high levels of viral oncogene transcripts in cervical carcinogenesis. Virus Res. 2011, 161, 124–130. [Google Scholar] [CrossRef]
- Tornesello, M.L.; Buonaguro, L.; Rossi, P.G.; Buonaguro, F.M. Viral and Cellular Biomarkers in the Diagnosis of Cervical Intraepithelial Neoplasia and Cancer. BioMed Res. Int. 2013, 2013, 1–10. [Google Scholar] [CrossRef]
- Cheung, J.L.; Cheung, T.-H.; Yu, M.Y.; Chan, P.K. Virological characteristics of cervical cancers carrying pure episomal form of HPV16 genome. Gynecol. Oncol. 2013, 131, 374–379. [Google Scholar] [CrossRef]
- McBride, A.A.; Warburton, A. The role of integration in oncogenic progression of HPV-associated cancers. PLOS Pathog. 2017, 13, e1006211. [Google Scholar] [CrossRef]
- Leung, T.-W.; Liu, S.S.; Leung, R.C.; Chu, M.M.; Cheung, A.N.; Ngan, H.Y.S. HPV 16 E2 binding sites 1 and 2 become more methylated than E2 binding site 4 during cervical carcinogenesis. J. Med. Virol. 2015, 87, 1022–1033. [Google Scholar] [CrossRef] [PubMed]
- Balderas-Loaeza, A.; Anaya-Saavedra, G.; Ramirez-Amador, V.A.; Guido-Jimenez, M.C.; Kalantari, M.; Calleja-Macias, I.E.; Bernard, H.-U.; García-Carrancá, A. Human papillomavirus-16 DNA methylation patterns support a causal association of the virus with oral squamous cell carcinomas. Int. J. Cancer 2007, 120, 2165–2169. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Sun, C.; Ghosh, A.; Rodriguez, A.C.; Schiffman, M.; Wentzensen, N.; Hildesheim, A.; Herrero, R.; Wacholder, S.; Lorincz, A.; et al. Methylation of Human Papillomavirus Type 16 Genome and Risk of Cervical Precancer in a Costa Rican Population. J. Natl. Cancer Inst. 2012, 104, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Wentzensen, N.; Sun, C.; Ghosh, A.; Kinney, W.; Mirabello, L.; Wacholder, S.; Shaber, R.; LaMere, B.; Clarke, M.; Lorincz, A.T.; et al. Methylation of HPV18, HPV31, and HPV45 Genomes and Cervical Intraepithelial Neoplasia Grade 3. J. Natl. Cancer Inst. 2012, 104, 1738–1749. [Google Scholar] [CrossRef]
- Yeo-Teh, N.S.L.; Ito, Y.; Jha, S. High-Risk Human Papillomaviral Oncogenes E6 and E7 Target Key Cellular Pathways to Achieve Oncogenesis. Int. J. Mol. Sci. 2018, 19, 1706. [Google Scholar] [CrossRef]
- Klymenko, T.; Hernandez-Lopez, H.; Macdonald, A.I.; Bodily, J.M.; Graham, S. Human Papillomavirus E2 Regulates SRSF3 (SRp20) To Promote Capsid Protein Expression in Infected Differentiated Keratinocytes. J. Virol. 2016, 90, 5047–5058. [Google Scholar] [CrossRef]
- Ramdan, A. A Study on Interactions between Different Viruses in the Pathogenesis of Cervical Cancer. Ph.D. Thesis, Univer-sity of Manchester, Manchester, UK, 2018. [Google Scholar]
- Yusupov, A.; Popovsky, D.; Mahmood, L.; Kim, A.S.; Akman, A.E.; Yuan, H. The nonavalent vaccine: A review of high-risk HPVs and a plea to the CDC. Am. J. Stem Cells 2019, 8, 52–64. [Google Scholar]
- Ciavattini, A.; Giannella, B.; De Vincenzo, R.; Di Giuseppe, J.; Papiccio, M.; Lukic, A.; Carpini, G.D.; Perino, A.; Frega, A.; Sopracordevole, F.; et al. HPV Vaccination: The Position Paper of the Italian Society of Colposcopy and Cervico-Vaginal Pathology (SICPCV). Vaccines 2020, 8, 354. [Google Scholar] [CrossRef]
- Wheeler, C.M.; Castellsagué, X.; Garland, S.M.; Szarewski, A.; Paavonen, J.; Naud, P.; Salmerón, J.; Chow, S.N.; Apter, D.; Kitchener, H.; et al. Cross-protective efficacy of HPV-16/18 AS04-adjuvanted vaccine against cervical infection and precancer caused by non-vaccine oncogenic HPV types: 4-year end-of-study analysis of the randomised, double-blind PATRICIA trial. Lancet Oncol. 2012, 13, 100–110. [Google Scholar] [CrossRef]
- Ault, K.A. Human papillomavirus vaccines and the potential for cross-protection between related HPV types. Gynecol. Oncol. 2007, 107 (Suppl. 1), S31–S33. [Google Scholar] [CrossRef]
- Toft, L.; Tolstrup, M.; Müller, M.; Sehr, P.; Bonde, J.; Storgaard, M.; Østergaard, L.; Søgaard, O.S. Comparison of the im-munogenicity of Cervarix® and Gardasil® human papillomavirus vaccines for oncogenic non-vaccine serotypes HPV-31, HPV-33, and HPV-45 in HIV-infected adults. Hum. Vaccines Immunother. 2014, 10, 1147–1154. [Google Scholar] [CrossRef] [PubMed]
- Herrero, R.; Wacholder, S.; Rodríguez, A.C.; Solomon, D.; González, P.; Kreimer, A.R.; Porras, C.; Schussler, J.; Jiménez, S.; Sherman, M.E.; et al. Prevention of persistent human papil-lomavirus infection by an HPV16/18 vaccine: A community-based randomized clinical trial in Guanacaste, Costa Rica. Cancer Discov. 2011, 1, 408–419. [Google Scholar] [CrossRef]
- Arbyn, M.; Xu, L. Efficacy and safety of prophylactic HPV vaccines. A Cochrane review of randomized trials. Expert Rev. Vaccines 2018, 17, 1085–1091. [Google Scholar] [CrossRef] [PubMed]
- Gravitt, P.E.; Winer, R.L. Natural History of HPV Infection across the Lifespan: Role of Viral Latency. Viruses 2017, 9, 267. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.; Dondog, B.; Waterboer, T.; Pawlita, M.; Tommasino, M.; Gheit, T. Abundance of multiple high-risk human pap-illomavirus (HPV) infections found in cervical cells analyzed by use of an ultrasensitive HPV genotyping assay. J. Clin. Microbiol. 2010, 48, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Hudelist, G.; Manavi, M.; Pischinger, K.I.; Watkins-Riedel, T.; Singer, C.F.; Kubista, E.; Czerwenka, K.F. Physical state and expression of HPV DNA in benign and dysplastic cervical tissue: Different levels of viral integration are correlated with lesion grade. Gynecol. Oncol. 2004, 92, 873–880. [Google Scholar] [CrossRef]
- Evans, B.A.; Bond, R.A.; Macrae, K.D. A colposcopic case-control study of cervical squamous intraepithelial lesions in women with anogenital warts. Sex. Transm. Infect. 1992, 68, 300–304. [Google Scholar] [CrossRef][Green Version]
- Silins, I.; Wang, Z.; Lundqvist, E.; Åvall-Frankendal, B.; Vikmanis, U.; Sapp, M.; Schiller, J.T.; Dillner, J. Serological evidence for protection by human papillomavirus (HPV) type 6 infection against HPV type 16 cervical carcinogenesis. J. Gen. Virol. 1999, 80, 2931–2936. [Google Scholar] [CrossRef][Green Version]
- Luostarinen, T.; Geijersstam, V.; Bjørge, T.; Eklund, C.; Hakama, M.; Hakulinen, T.; Jellum, E.; Koskela, P.; Paavonen, J.; Pukkala, E.; et al. No excess risk of cervical carcinoma among women seropositive for both HPV16 and HPV6/11. Int. J. Cancer 1999, 80, 818–822. [Google Scholar] [CrossRef]
- Luostarinen, T.; Lehtinen, M.; Bjørge, T.; Abeler, V.; Hakama, M.; Hallmans, G.; Jellum, E.; Koskela, P.; Lenner, P.; Lie, A.K.; et al. Joint effects of dif-ferent human papillomaviruses and Chlamydia trachomatis infections on risk of squamous cell carcinoma of the cervix uteri. Eur. J. Cancer 2004, 40, 1058–1065. [Google Scholar] [CrossRef]
- Sundström, K.; Ploner, A.; Arnheim-Dahlström, L.; Eloranta, S.; Palmgren, J.; Adami, H.-O.; Helm, N.Y.; Sparén, P.; Dillner, J. Interactions Between High- and Low-Risk HPV Types Reduce the Risk of Squamous Cervical Cancer. J. Natl. Cancer Inst. 2015, 107, 10. [Google Scholar] [CrossRef] [PubMed]
- Maranga, I.O.; Hampson, L.; Oliver, A.W.; He, X.; Gichangi, P.; Rana, F.; Opiyo, A.; Hampson, I.N. HIV Infection Alters the Spectrum of HPV Subtypes Found in Cervical Smears and Carcinomas from Kenyan Women. Open Virol. J. 2013, 7, 19–27. [Google Scholar] [CrossRef]
- E McLaughlin-Drubin, M.; Meyers, C. Evidence for the coexistence of two genital HPV types within the same host cell in vitro. Virology 2004, 321, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Kusumoto-Matsuo, R.; Ishii, Y.; Takeuchi, T.; Kukimoto, I. Replication interference between human papillomavirus types 16 and 18 mediated by heterologous E1 helicases. Virology 2014, 11, 11. [Google Scholar] [CrossRef] [PubMed]
- Biryukov, J.; Meyers, C. Superinfection Exclusion between Two High-Risk Human Papillomavirus Types during a Coinfec-tion. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Callegari, E.T.; Tabrizi, S.; Pyman, J.; Saville, M.; Cornall, A.M.; Brotherton, J.M.; Garland, S.M. How best to interpret mixed human papillomavirus genotypes in high-grade cervical intraepithelial neoplasia lesions. Vaccine 2014, 32, 4082–4088. [Google Scholar] [CrossRef] [PubMed]
- van der Marel, J.; Berkhof, J.; Ordi, J.; Torné, A.; Del Pino, M.; van Baars, R.; Schiffman, M.; Wentzensen, N.; Jenkins, D.; Quint, W.G. Attributing oncogenic human papillomavirus genotypes to high-grade cervical neoplasia: Which type causes the lesion? Am. J. Surg. Pathol. 2015, 39, 496–504. [Google Scholar] [CrossRef]
- Venetianer, R.; Clarke, M.A.; Van Der Marel, J.; Tota, J.; Schiffman, M.; Dunn, S.T.; Walker, J.; Zuna, R.; Quint, W.; Wentzensen, N. Identification of HPV Genotypes Causing Cervical Pre-Cancer using Tissue-Based Genotyping. Int. J. Cancer 2020, 146, 2836–2844. [Google Scholar] [CrossRef]
- Shen, Z.; Liu, X.; Morihara, J.; Hulbert, A.; Koutsky, L.A.; Kiviat, N.B.; Xi, L.F. Detection of Human Papillomavirus Infec-tions at the Single-Cell Level. Intervirology 2015, 58, 324–331. [Google Scholar] [CrossRef]
- Hammer, A.; De Koning, M.N.; Blaakaer, J.; Steiniche, T.; Doorbar, J.; Griffin, H.; Mejlgaard, E.; Svanholm, H.; Quint, W.G.; Gravitt, P.E. Whole tissue cervical mapping of HPV infection: Molecular evidence for focal latent HPV infection in humans. Papillomavirus Res. 2019, 7, 82–87. [Google Scholar] [CrossRef]
- Van Schalkwyk, C.; Moodley, J.; Welte, A.; Johnson, L.F. Estimated impact of human papillomavirus vaccines on infection burden: The effect of structural assumptions. Vaccine 2019, 37, 5460–5465. [Google Scholar] [CrossRef] [PubMed]
- Alizon, S.; Murall, C.L.; Bravo, I.G. Why Human Papillomavirus Acute Infections Matter. Viruses 2017, 9, 293. [Google Scholar] [CrossRef] [PubMed]
- Orlando, P.A.; Gatenby, R.A.; Giuliano, A.R.; Brown, J.S. Evolutionary Ecology of Human Papillomavirus: Trade-offs, Coexistence, and Origins of High-Risk and Low-Risk Types. J. Infect. Dis. 2012, 205, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Murall, C.L.; Bauch, C.T.; Day, T. Could the human papillomavirus vaccines drive virulence evolution? Proc. Biol. Sci. 2015, 282, 20141069. [Google Scholar] [CrossRef] [PubMed]
- Alizon, S.; Murall, C.L.; Saulnier, E.; Sofonea, M.T. Detecting within-host interactions from genotype combination prevalence data. Epidemics 2019, 29, 100349. [Google Scholar] [CrossRef] [PubMed]
- MIller, N. Clinical Review of Biologics License Application for Human Papillomavirus 6, 11, 16, 18 L1 Virus Like Particle Vaccine (S. cerevisiae) (STN 125126 GARDASIL); Merck, Inc.: Kenilworth, NJ, USA, 2006. [Google Scholar]
- European Medicines Agency. Assessment Report for Gardasil; European Medicines Agency: London, UK, 2008. [Google Scholar]
- Carozzi, F.M.; Tornesello, M.L.; Burroni, E.; Loquercio, G.; Carillo, G.; Angeloni, C.; Scalisi, A.; Macis, R.; Chini, F.; Buonaguro, F.M.; et al. Prevalence of Human Papillomavirus Types in High-Grade Cervical Intraepithelial Neoplasia and Cancer in Italy. Cancer Epidemiol. Biomark. Prev. 2010, 19, 2389–2400. [Google Scholar] [CrossRef]
- De Martel, C.; Plummer, M.; Vignat, J.; Franceschi, S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int. J. Cancer 2017, 141, 664–670. [Google Scholar] [CrossRef]
- Mesher, D.; Soldan, K.; Lehtinen, M.; Beddows, S.; Brisson, M.; Brotherton, J.M.; Chow, E.P.; Cummings, T.; Drolet, M.; Fairley, C.K.; et al. Population-Level Effects of Human Papillomavirus Vaccination Pro-grams on Infections with Nonvaccine Genotypes. Emerg. Infect. Dis. 2016, 22, 1732–1740. [Google Scholar] [CrossRef]
- Gray, P.; Palmroth, J.; Luostarinen, T.; Apter, D.; Dubin, G.; Garnett, G.; Eriksson, T.; Natunen, K.; Merikukka, M.; Pimenoff, V.; et al. Evaluation of HPV type-replacement in unvaccinated and vaccinated adolescent females-Post-hoc analysis of a community-randomized clinical trial (II). Int. J. Cancer 2018, 142, 2491–2500. [Google Scholar] [CrossRef]
- Gray, P.; Luostarinen, T.; Vänskä, S.; Eriksson, T.; Lagheden, C.; Man, I.; Palmroth, J.; Pimenoff, V.N.; Söderlund-Strand, A.; Dillner, J.; et al. Occurrence of human papillomavirus (HPV) type replacement by sexual risk-taking behaviour group: Post-hoc analysis of a community randomized clinical trial up to nine years after vaccination (IV). Int. J. Cancer 2019, 145, 785–796. [Google Scholar] [CrossRef]
- Man, I.; Vänskä, S.; Lehtinen, M.; Bogaards, J.A. Human Papillomavirus Genotype Replacement: Still Too Early to Tell? J. Infect. Dis. 2020. [Google Scholar] [CrossRef] [PubMed]
- McClung, N.M.; Gargano, J.W.; Park, I.U.; Whitney, E.; Abdullah, N.; Ehlers, S.; Bennett, N.M.; Scahill, M.; Niccolai, L.M.; Brackney, M.; et al. Estimated Number of Cases of High-Grade Cervical Lesions Diagnosed Among Women—United States, 2008 and 2016. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 337–343. [Google Scholar] [CrossRef] [PubMed]
- Innes, C.R.; Sykes, P.H.; Harker, D.; Williman, J.A.; Van der Griend, R.A.; Whitehead, M.; Hibma, M.; Lawton, B.A.; Fitz-gerald, P.; Dudley, N.M.; et al. Changes in human papillo-mavirus genotypes associated with cervical intraepithelial neoplasia grade 2 lesions in a cohort of young women (2013–2016). Papillomavirus Res. 2018, 6, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Innes, C.R.; Williman, J.A.; Simcock, B.J.; Hider, P.; Sage, M.; Dempster-Rivett, K.; Lawton, B.A.; Sykes, P.H. Impact of human papillomavirus vaccination on rates of abnormal cervical cytology and histology in young New Zealand women. New Zealand Med. J. 2020, 133, 72–84. [Google Scholar]
- Kang, Y.-J.; Lewis, H.; Smith, M.A.; Simonella, L.; Neal, H.; Bromhead, C.; Canfell, K. Pre-vaccination type-specific HPV prevalence in confirmed cervical high grade lesions in the Māori and non-Māori populations in New Zealand. BMC Infect. Dis. 2015, 15, 365. [Google Scholar] [CrossRef]
- Drolet, M.; Bénard, É.; Pérez, N.; Brisson, M.; HPV Vaccination Impact Study Group. Population-level impact and herd effects following the introduction of human papillomavirus vaccination programmes: Updated systematic review and meta-analysis. Lancet 2019, 394, 497–509. [Google Scholar] [CrossRef]
- Harper, D.M.; Demars, L.R. HPV vaccines—A review of the first decade. Gynecol. Oncol. 2017, 146, 196–204. [Google Scholar] [CrossRef]
- Ryser, M.; Berlaimont, V.; Karkada, N.; Mihalyi, A.; Rappuoli, R.; Van Der Most, R.G. Post-hoc analysis from phase III trials of human papillomavirus vaccines: Considerations on impact on non-vaccine types. Expert Rev. Vaccines 2019, 18, 309–322. [Google Scholar] [CrossRef]
- Rees, C.; Brhlikova, P.; Pollock, A.M. Will HPV vaccination prevent cervical cancer? J. R. Soc. Med. 2020, 113, 64–78. [Google Scholar] [CrossRef]
- Ährlund-Richter, A.; Cheng, L.; Hu, Y.O.O.; Svensson, M.; Pennhag, A.A.L.; Ursu, R.G.; Haeggblom, L.; Grün, N.; Ramqvist, T.; Engstrand, L.; et al. Changes in Cervical Human Papillomavirus (HPV) Prevalence at a Youth Clinic in Stockholm, Sweden, a Decade After the Introduction of the HPV Vaccine. Front. Cell. Infect. Microbiol. 2019, 9, 59. [Google Scholar] [CrossRef]
- Arbyn, M.; Weiderpass, E.; Bruni, L.; De Sanjosé, S.; Saraiya, M.; Ferlay, J.; Bray, F. Estimates of incidence and mortality of cervical cancer in 2018: A worldwide analysis. Lancet Glob. Heal. 2020, 8, e191–e203. [Google Scholar] [CrossRef]
- CRUK Cervical Cancer Incidence Statistics. Available online: https://www.cancerresearchuk.org/health-professional/cancer-statistics/statistics-by-cancer-type/cervical-cancer/incidence#heading-Two (accessed on 1 September 2020).
- Australian-Government. Cancer Incidence. In National Cancer Control Indicarors; Australian-Government: Strawberry Hills, NSW, Australia, 2019. [Google Scholar]
- O’Hallahan, M.C.H.; Sage, M. Clinical Practice Guidelines for Cervical Screening in New Zealand 2020 Draft; National Screeing Unit, Minsitry of Health: Wellington, New Zealand, 2020. [Google Scholar]
- Wang, J.; Andrae, B.; Strander, B.; Sparén, P.; Dillner, J. Increase of cervical cancer incidence in Sweden in relation to screening history: Population cohort study. Acta Oncol. 2020, 59, 988–993. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Ploner, A.; Elfström, K.M.; Wang, J.; Roth, A.; Fang, F.; Sundström, K.; Dillner, J.; Sparén, P. HPV Vaccination and the Risk of Invasive Cervical Cancer. N. Engl. J. Med. 2020, 383, 1340–1348. [Google Scholar] [CrossRef] [PubMed]
- Orumaa, M.; Leinonen, M.K.; Campbell, S.; Møller, B.; Myklebust, T.A.; Nygard, M. Recent increase in incidence of cervical precancerous lesions in Norway: Nationwide study from 1992 to 2016. Int. J. Cancer 2019, 145, 2629–2638. [Google Scholar] [CrossRef]
- Hansen, B.T.; Campbell, S.; Nygård, M. Long-term incidence trends of HPV-related cancers, and cases preventable by HPV vaccination: A registry-based study in Norway. BMJ Open 2018, 8, e019005. [Google Scholar] [CrossRef]
- Murall, C.L.; Rahmoun, M.; Selinger, C.; Baldellou, M.; Bernat, C.; Bonneau, M.; Boué, V.; Buisson, M.; Christophe, G.; D’Auria, G.; et al. Natural history, dynamics, and ecology of human papillomaviruses in genital infections of young women: Protocol of the PAPCLEAR cohort study. BMJ Open 2019, 9, e025129. [Google Scholar] [CrossRef]
- Murall, C.L.; Reyné, B.; Selinger, C.; Bernat, C.; Boué, V.; Grasset, S.; Groc, S.; Rahmoun, M.; Bender, N.; Bonneau, M.; et al. HPV cervical infections and serological status in vaccinated and unvaccinated women. Vaccine 2020, 38, 8167–8174. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Disease | HPV Type |
|---|---|
| Plantar warts | 1, 2, 4, 63 |
| Common warts | 2, 1, 7, 4, 26, 27, 29, 41, 57, 65, 77, 3, 10, 28 |
| Flat warts | 3, 10, 26, 27, 28, 38, 41, 49, 75, 76 |
| Other cutaneous lesions (e.g., epidermoid cysts, laryngeal carcinoma) | 6, 11, 16, 30, 33, 36, 37, 38, 41, 48, 60, 72, 73 |
| Epidermodysplasia verruciformis | 2, 3, 10, 5, 8, 9, 12, 14, 15, 17, 19, 20, 21, 22, 23, 24, 25, 36, 37, 38, 47, 50 |
| Recurrent respiratory papillomatosis | 6, 11 |
| Focal epithelial hyperplasia of Heck | 13, 32 |
| Conjunctival papillomas/carcinomas | 6, 11, 16 |
| Condyloma acuminata (genital warts) | 6, 11, 30, 42, 43, 45, 51, 54, 55, 70 |
| Cervical intraepithelial neoplasia | |
| Unspecified | 30, 34, 39, 40, 53, 57, 59, 61, 62, 64, 66, 67, 68, 69 |
| Low risk | 6, 11, 16, 18, 31, 33, 35, 42, 43, 44, 45, 51, 52, 74 |
| High risk | 16, 18, 6, 11, 31, 34, 33, 35, 39, 42, 44, 45, 51, 52, 56, 58, 66 |
| Cervical carcinoma | 16, 18, 31, 45, 33, 35, 39, 51, 52, 56, 58, 66, 68, 70 |
| Gardasil N = 10268 | Placebo N = 10273 | ||||||
|---|---|---|---|---|---|---|---|
| Day 1 Status | N * | No. of Cases | Incidence Rate/100 Person Years at Risk | N * | No. of Cases | Incidence Rate/100 Person Years at Risk | Vaccine Efficacy 95% CI |
| MITT-3 | 9831 | 122 | 0.6 | 9896 | 201 | 0.9 | 39.0% (23.3, 51.7%) |
| PCR (-) Sero (-) | 9342 | 1 | 0.0 | 9400 | 81 | 0.4 | 98.8% (92.9, 100.0%) |
| PCR (-) Sero (+) | 853 | 0 | 0.0 | 910 | 4 | 0.2 | 100.0% (−63.6, 100.0%) |
| PCR (+) Sero (-) | 661 | 42 | 3.2 | 626 | 57 | 4.6 | 31.2% (−4.5, 54.9%) |
| PCR (+) Sero (+) | 473 | 79 | 9.1 | 499 | 69 | 7.3 | −25.8% (−76.4, 10.1%) |
| Sero and/or PCR (+) | (121) | (130) | (No efficacy estimate provided) | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hampson, I.N.; Oliver, A.W.; Hampson, L. Potential Effects of Human Papillomavirus Type Substitution, Superinfection Exclusion and Latency on the Efficacy of the Current L1 Prophylactic Vaccines. Viruses 2021, 13, 22. https://doi.org/10.3390/v13010022
Hampson IN, Oliver AW, Hampson L. Potential Effects of Human Papillomavirus Type Substitution, Superinfection Exclusion and Latency on the Efficacy of the Current L1 Prophylactic Vaccines. Viruses. 2021; 13(1):22. https://doi.org/10.3390/v13010022
Chicago/Turabian StyleHampson, Ian N., Anthony W. Oliver, and Lynne Hampson. 2021. "Potential Effects of Human Papillomavirus Type Substitution, Superinfection Exclusion and Latency on the Efficacy of the Current L1 Prophylactic Vaccines" Viruses 13, no. 1: 22. https://doi.org/10.3390/v13010022
APA StyleHampson, I. N., Oliver, A. W., & Hampson, L. (2021). Potential Effects of Human Papillomavirus Type Substitution, Superinfection Exclusion and Latency on the Efficacy of the Current L1 Prophylactic Vaccines. Viruses, 13(1), 22. https://doi.org/10.3390/v13010022