Optimization Rules for SARS-CoV-2 Mpro Antivirals: Ensemble Docking and Exploration of the Coronavirus Protease Active Site

 ,
,  ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Preparation of SARS-CoV-2 Mpro Receptors for Docking
2.2. Preparation of Ligands for Docking into SARS-CoV-2 Mpro
2.3. Docking Ligands into the SARS-CoV-2 Mpro Receptor
2.4. Structural Evaluation of SARS-CoV-2 Mpro Receptors
2.5. Conservation Analysis of SARS-CoV-2 Mpro Receptor
2.6. Calculation of Physiochemical Properties and Bioactivity
2.7. Molecular Dynamics of CM02, CM06, and CM07 Complex with SARS-CoV-2 Mpro Receptor (6LU7)
3. Results and Discussion
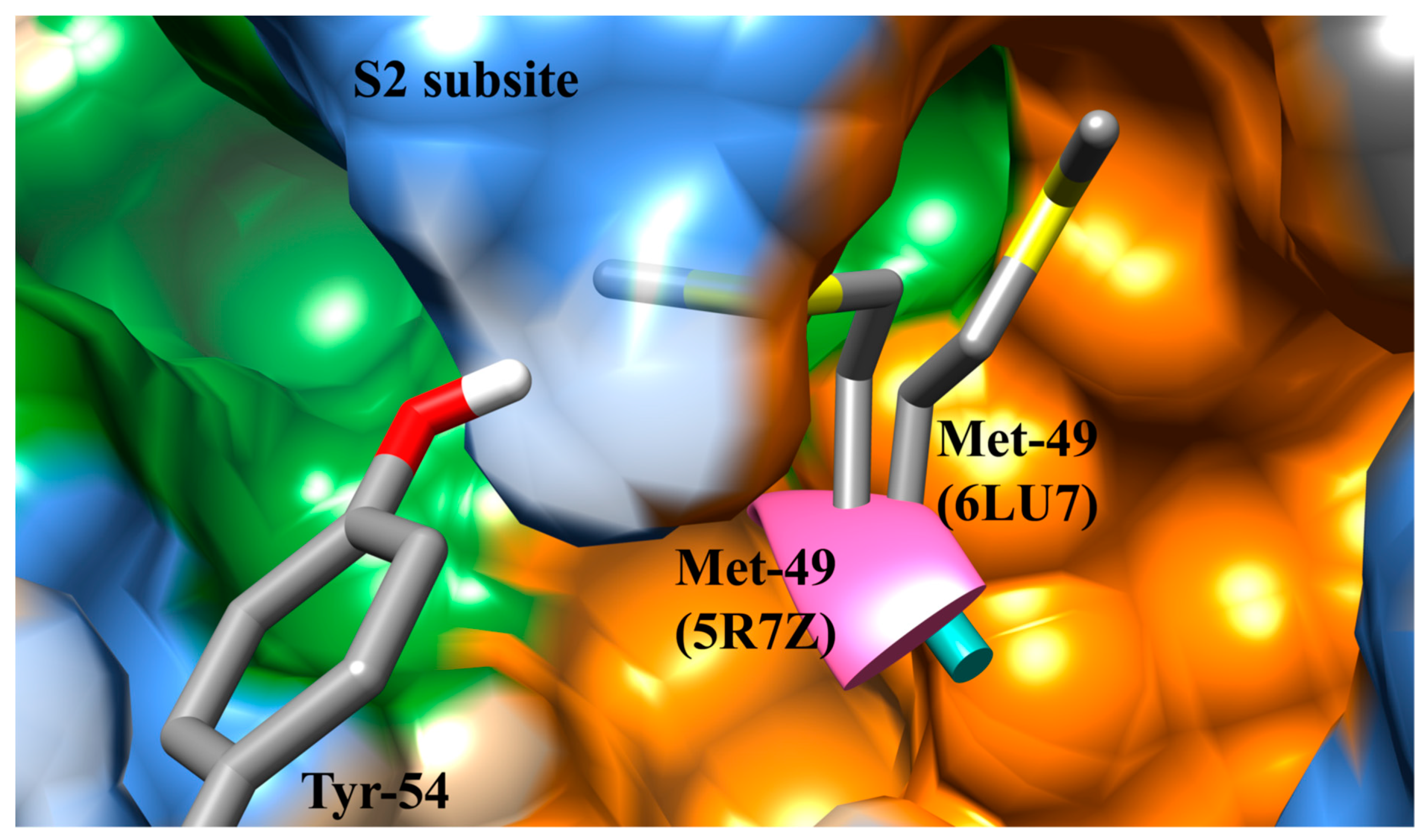
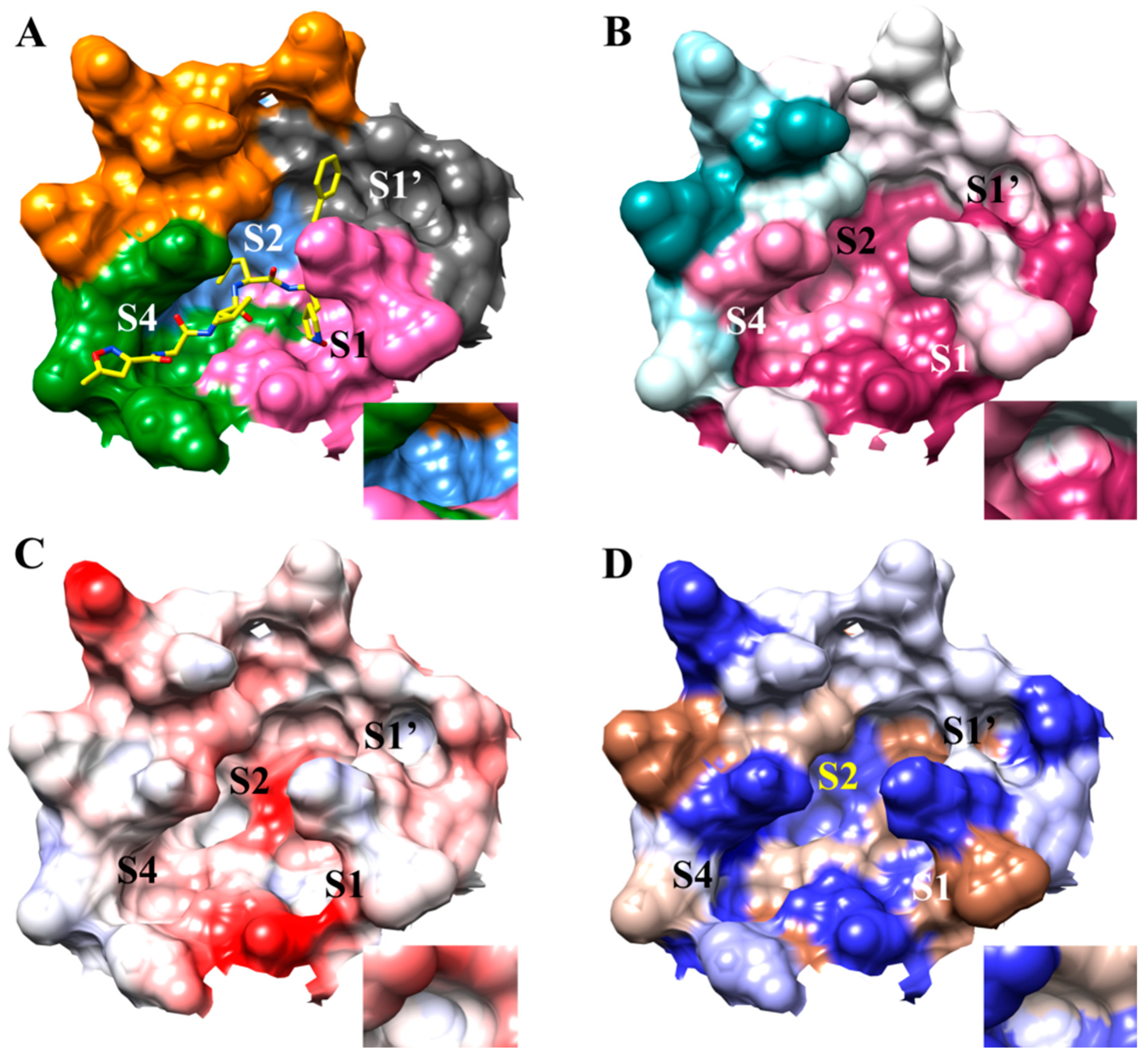
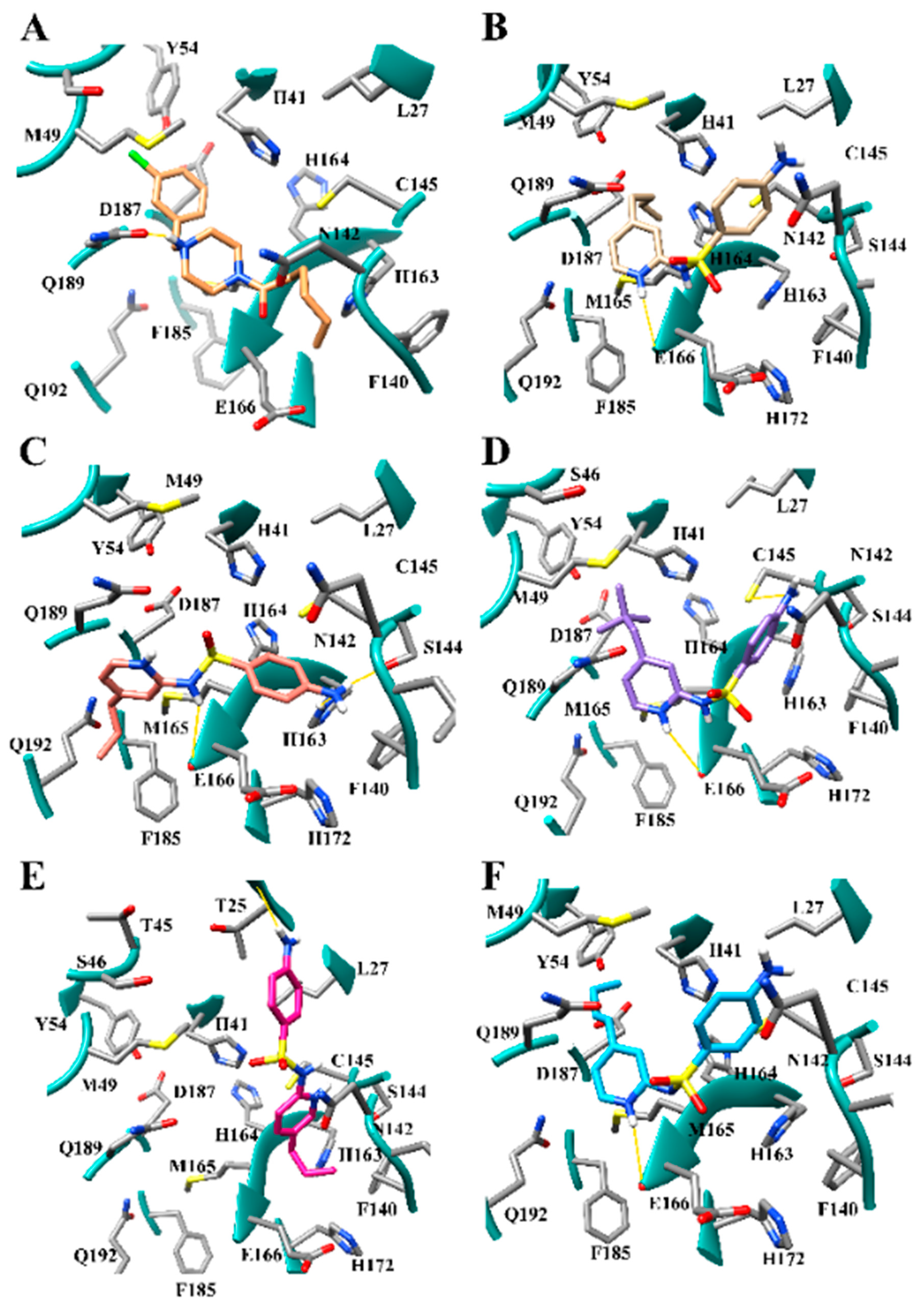
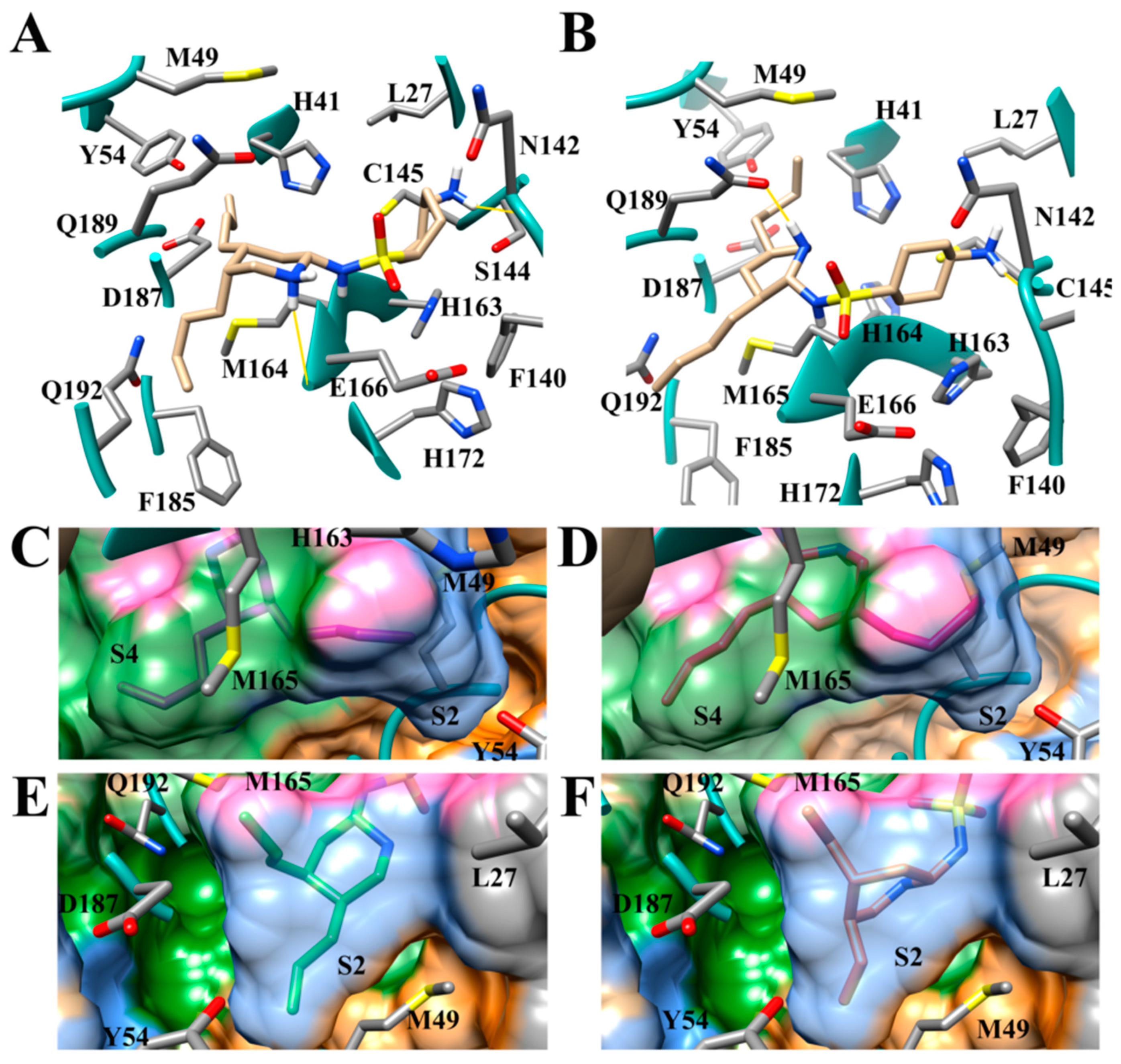
3.1. The SARS-CoV-2 Mpro Active Site
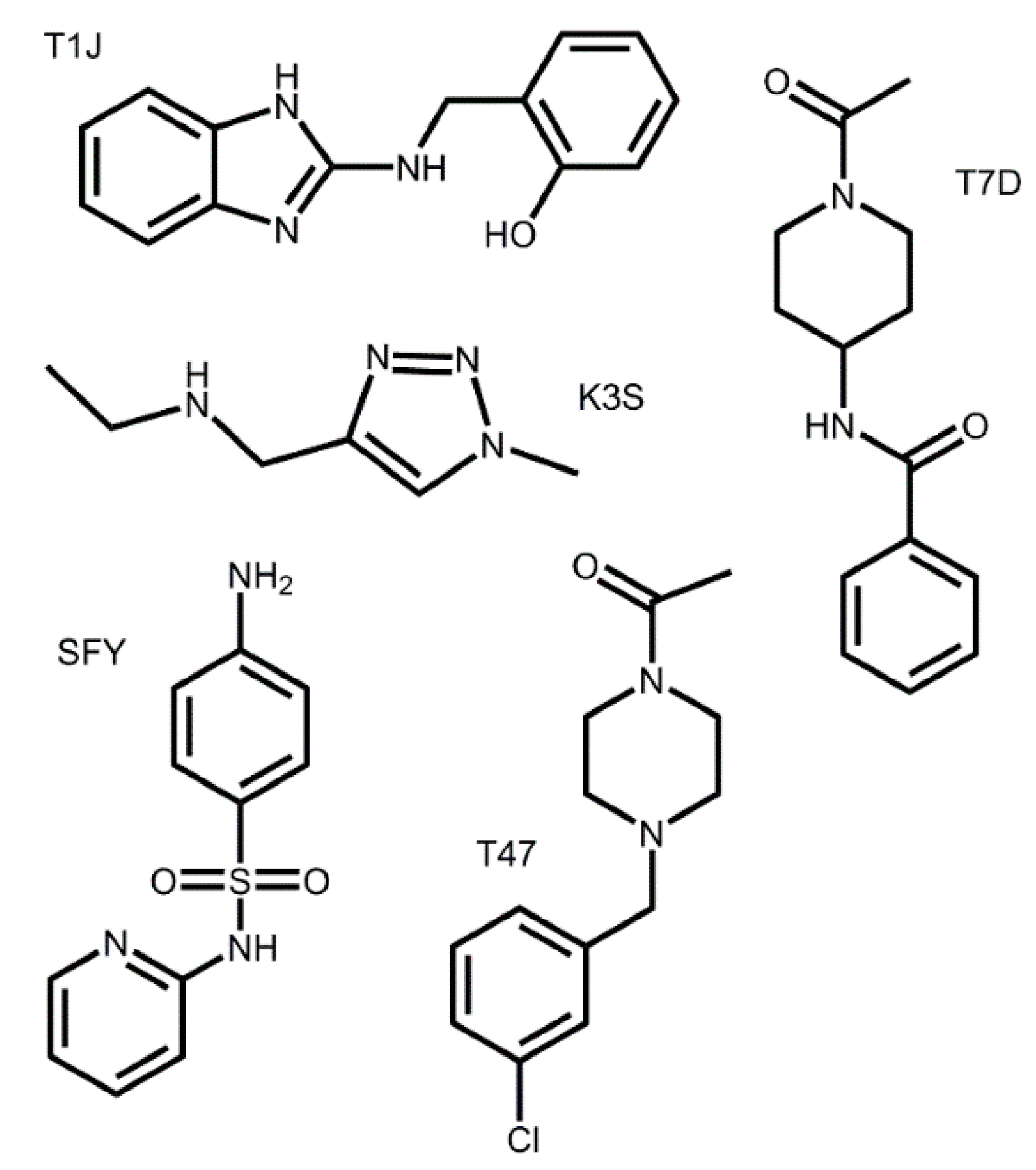
3.2. Docking of Bound Zinc Database Inhibitors in Coronavirus Receptors
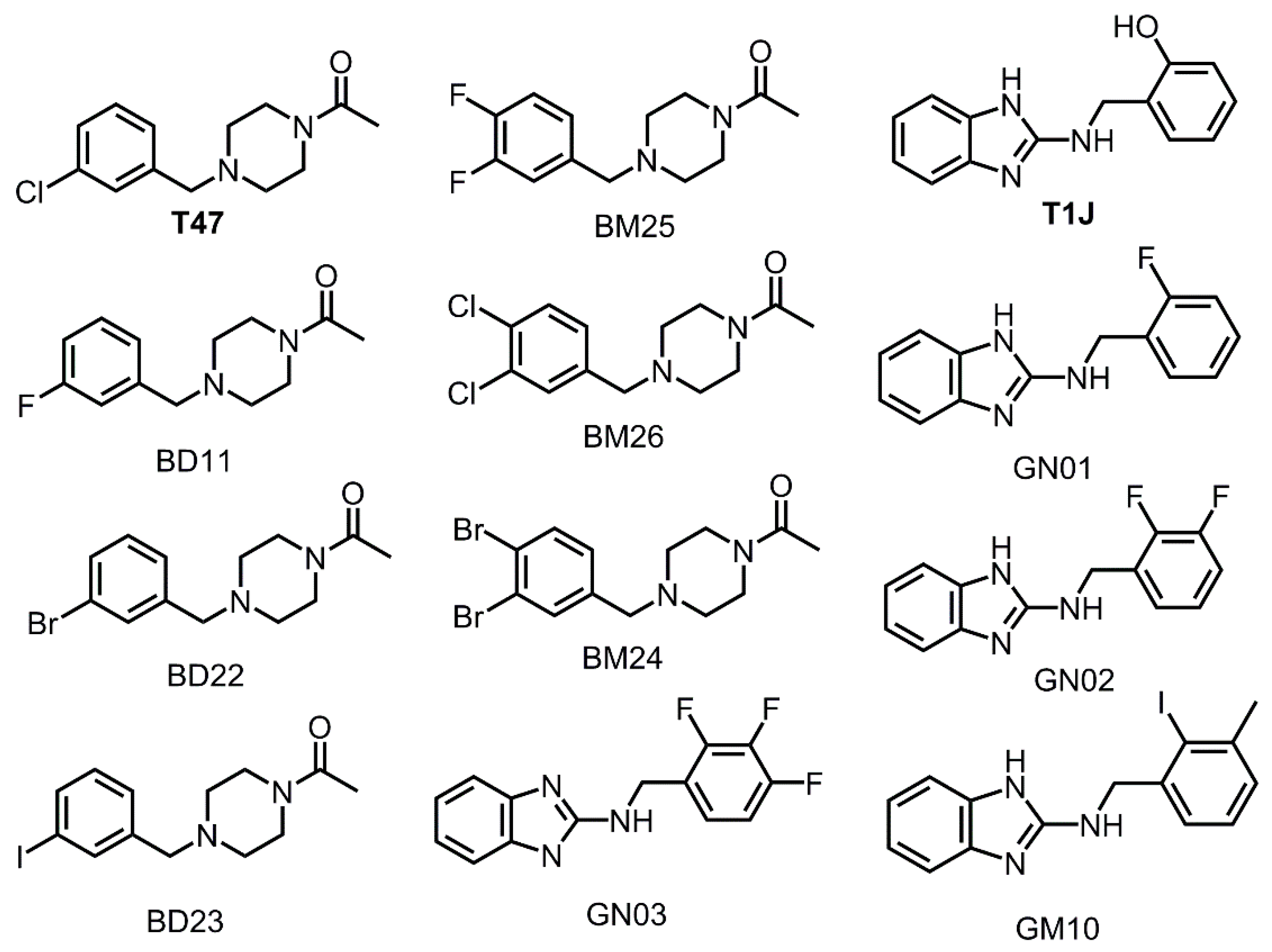
3.3. Halogens in the SARS-CoV-2 Mpro Actives Site Have Little Impact on Binding Affinity
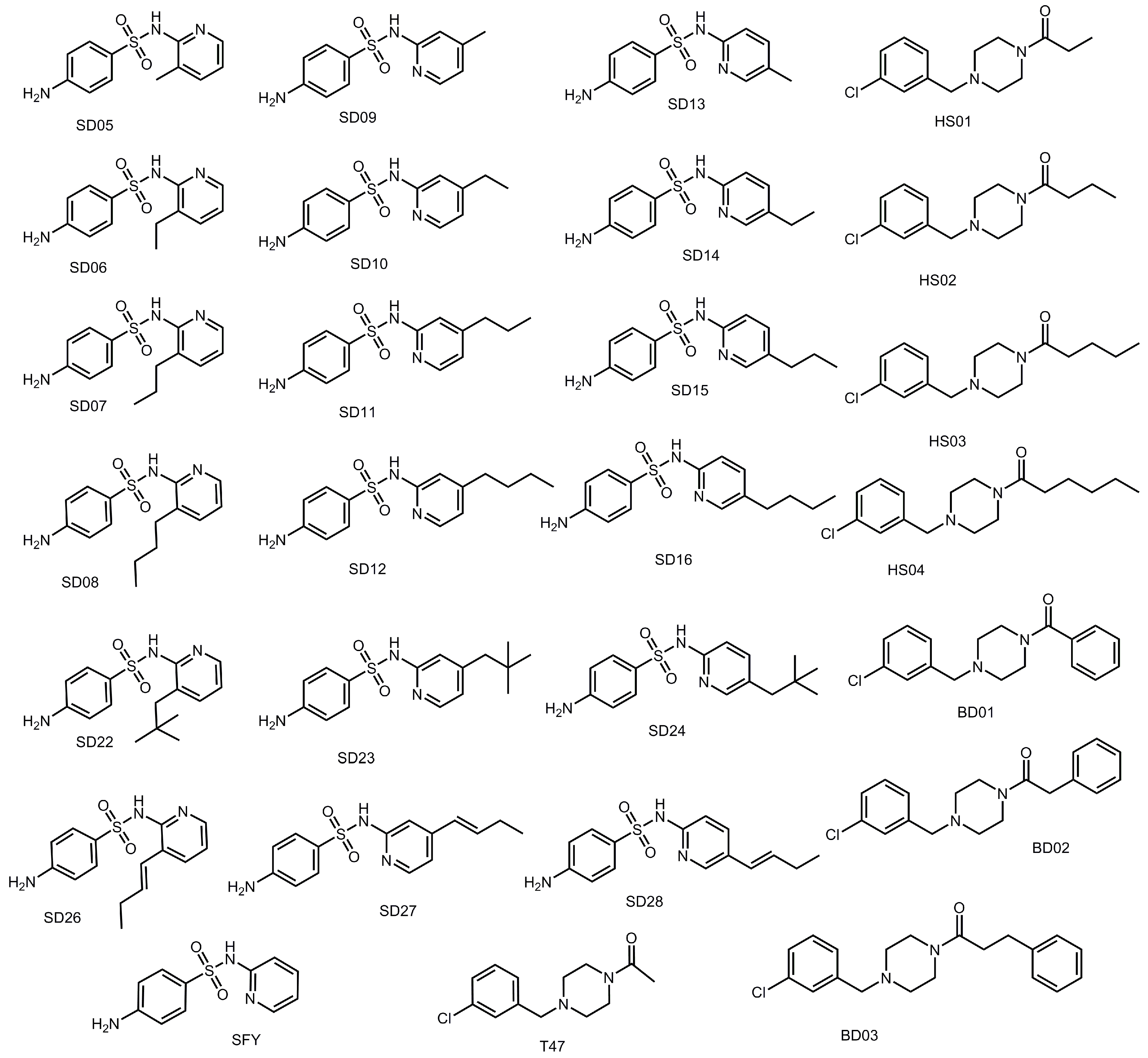
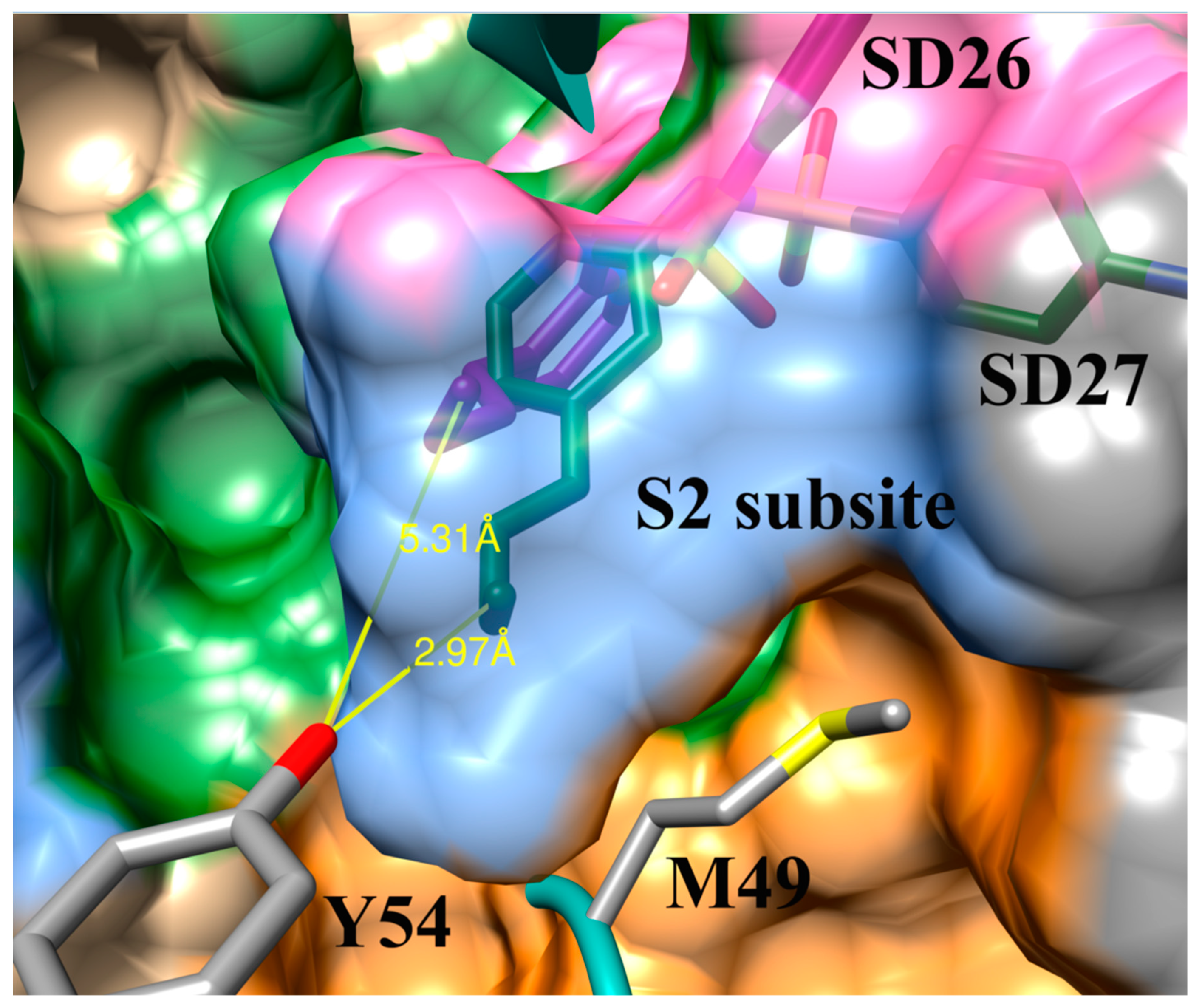
3.4. Addition of Aliphatic Substituents Increases Binding Affinity to SARS-CoV-2 Mpro Receptor
3.5. Nitrogen Heterocycles can Increase Binding Affinity to SARS-CoV-2 Active Site
3.6. Aliphatic Rings Improve Binding Afinity to SARS-CoV-2 Mpro Active Site
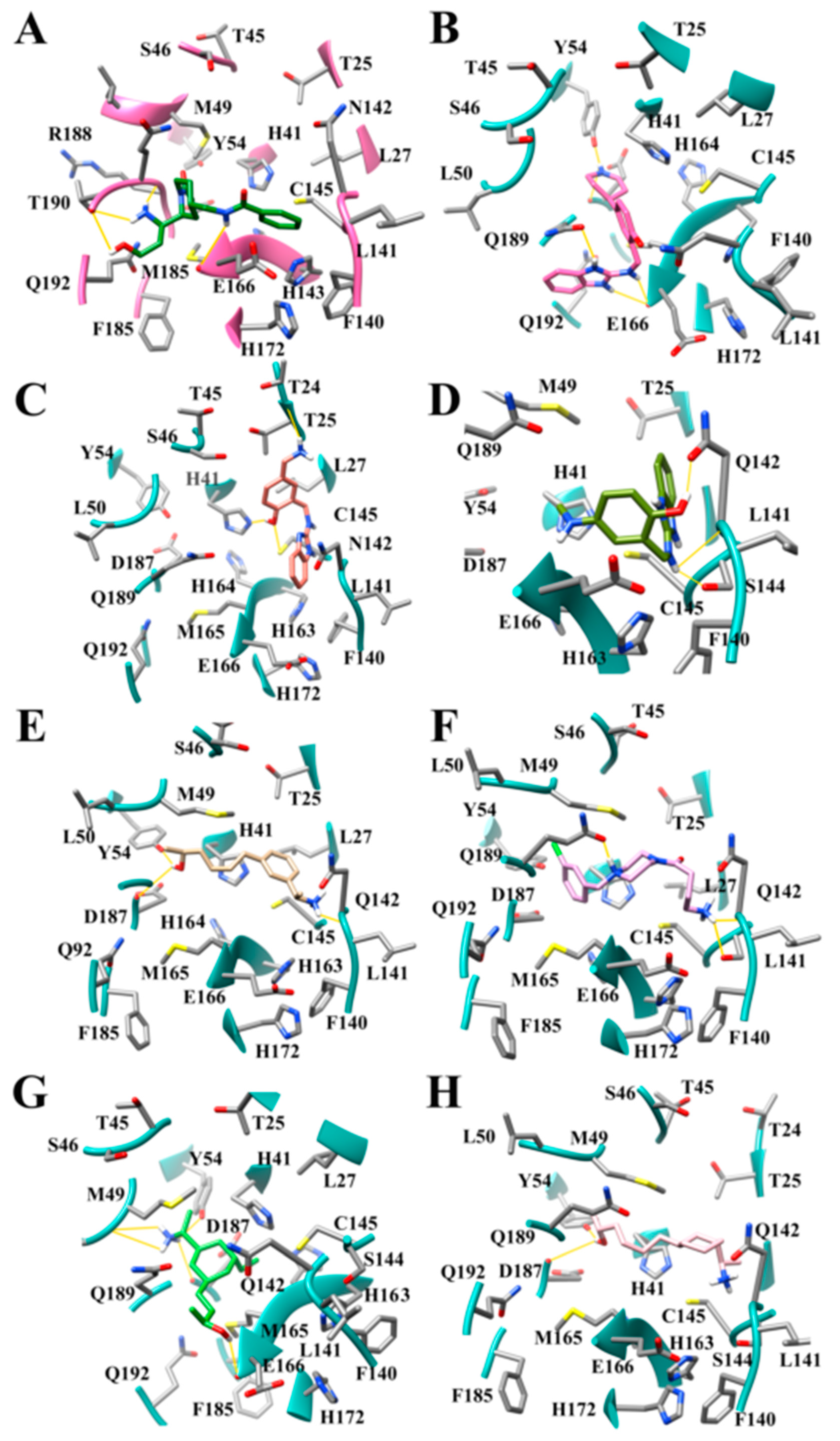
3.7. Several Hydrogen Bonding Hotspots are Present in SARS-CoV-2 Mpro
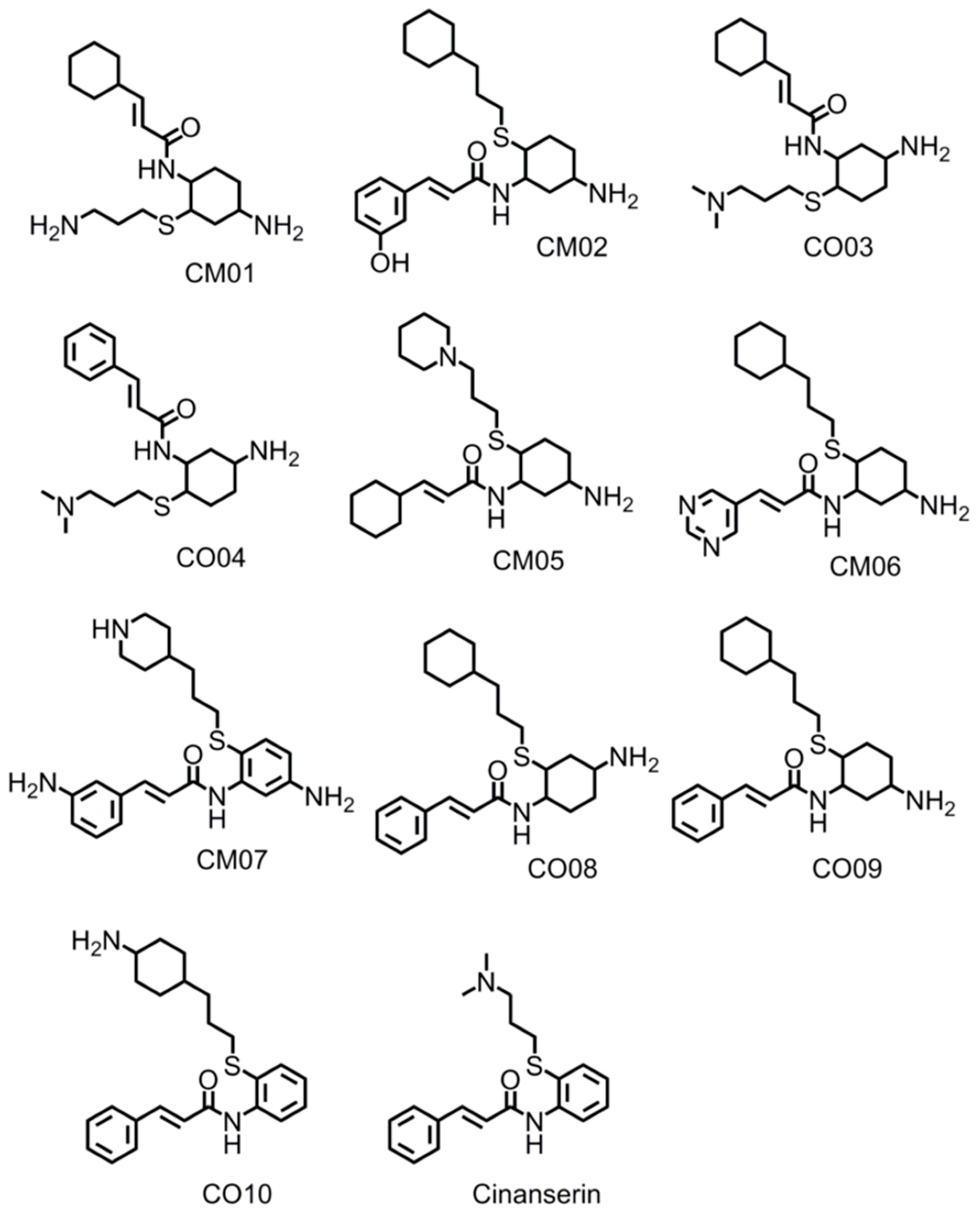
3.8. Design of Compounds with Optimized Binding Affinity to SARS-CoV-2 Mpro
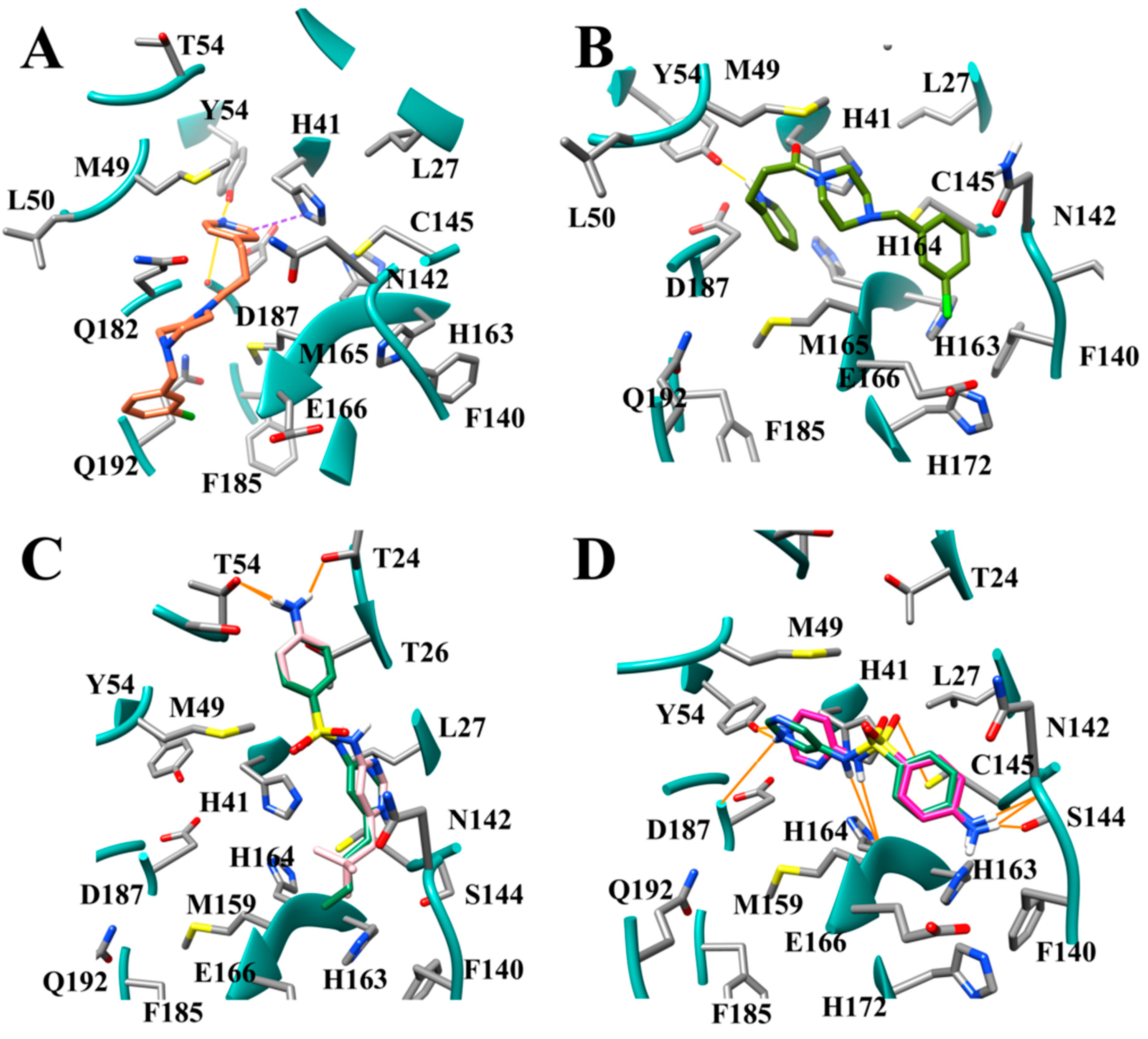
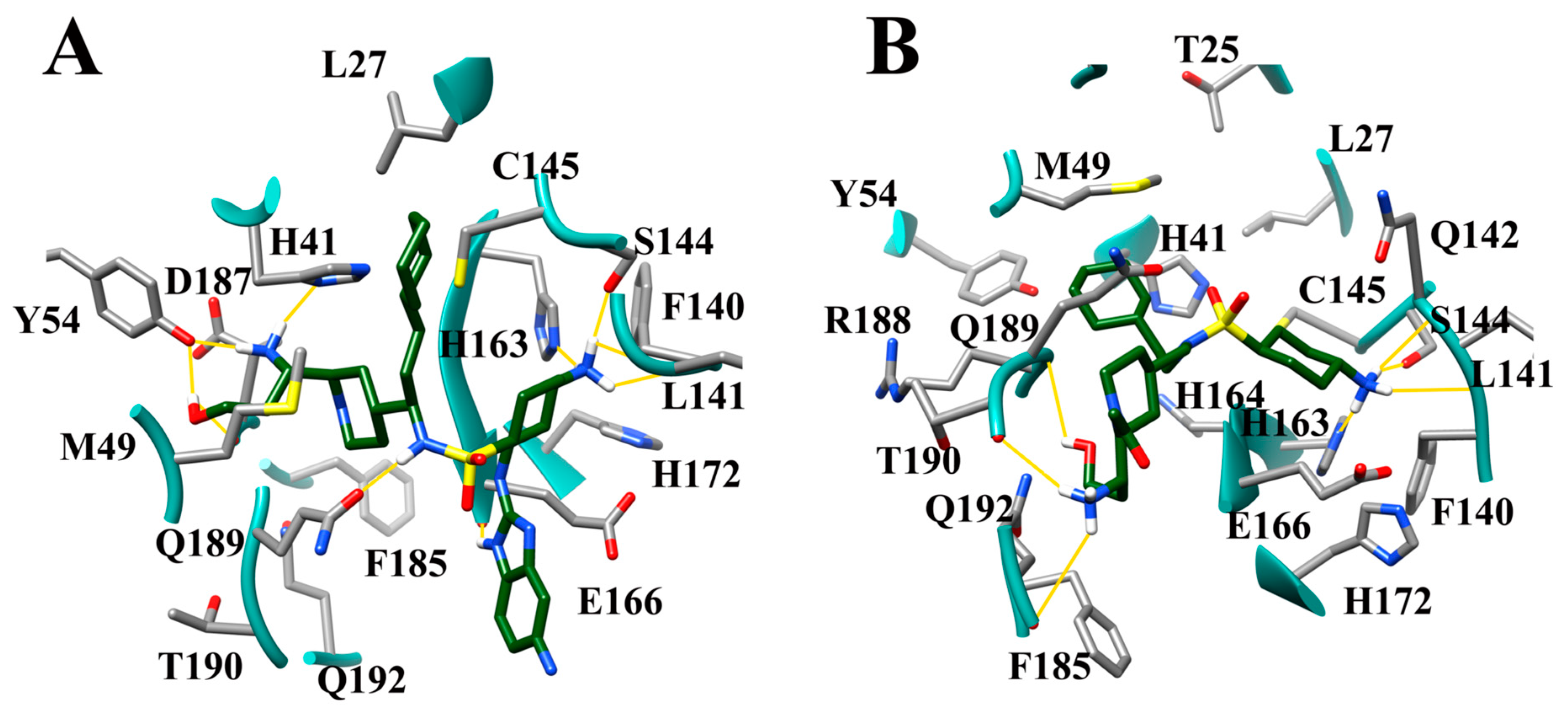
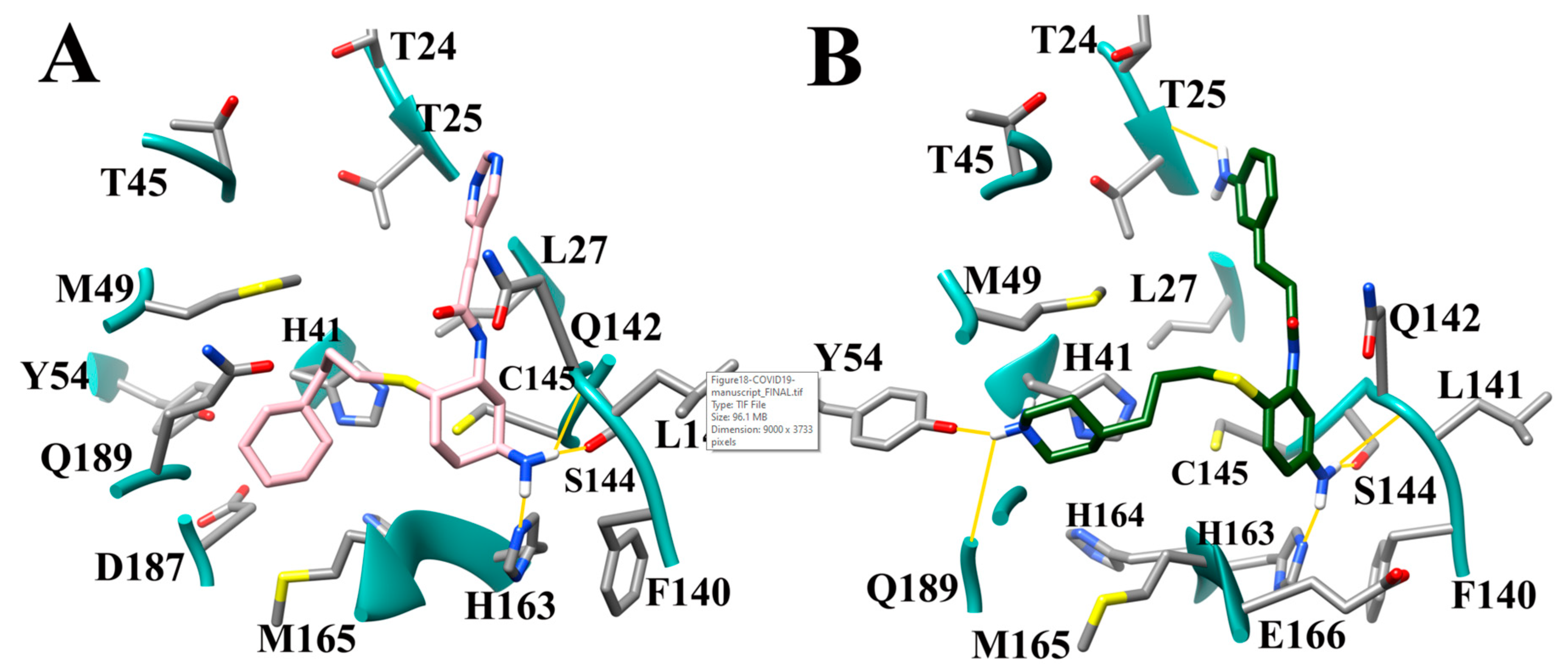
3.9. Optimization of Cinanserin Hit for SARS-CoV-2 Mpro
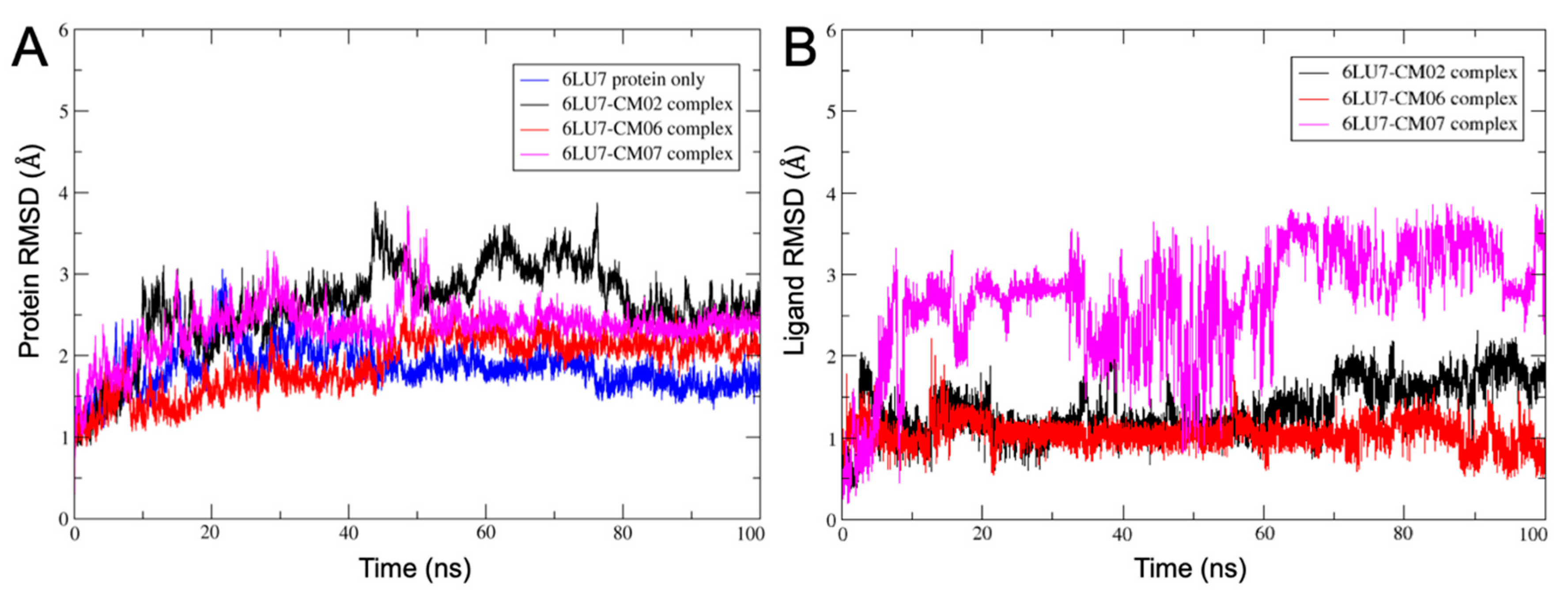
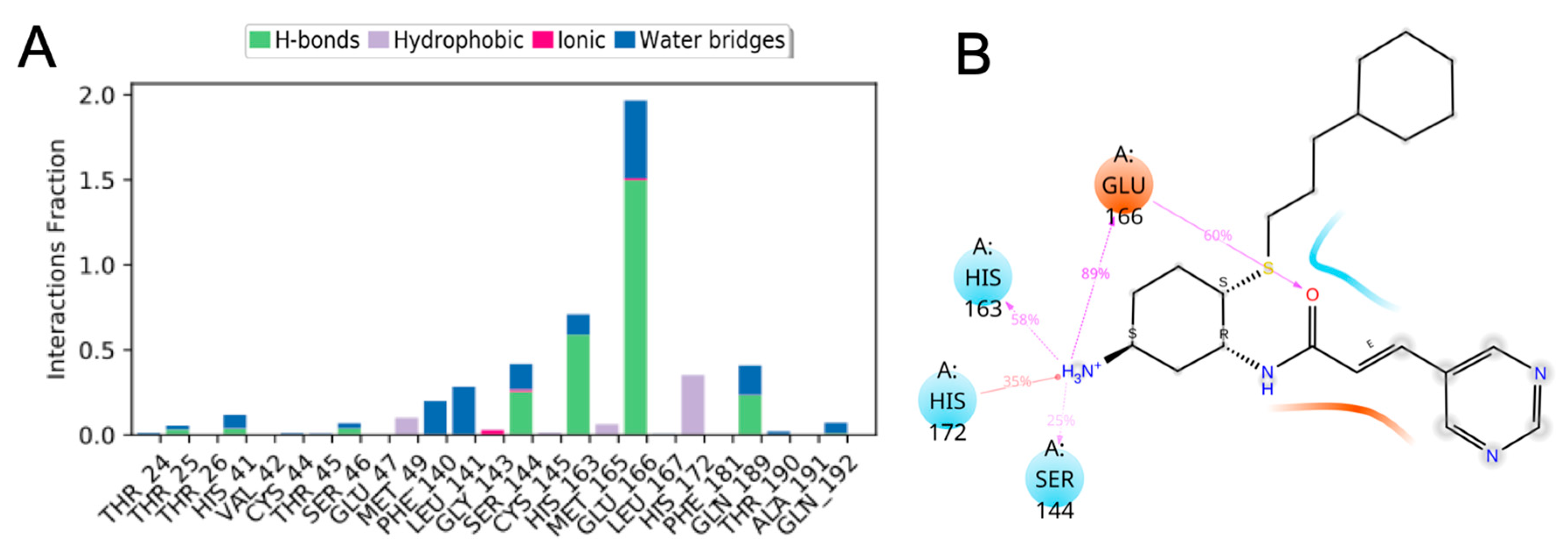
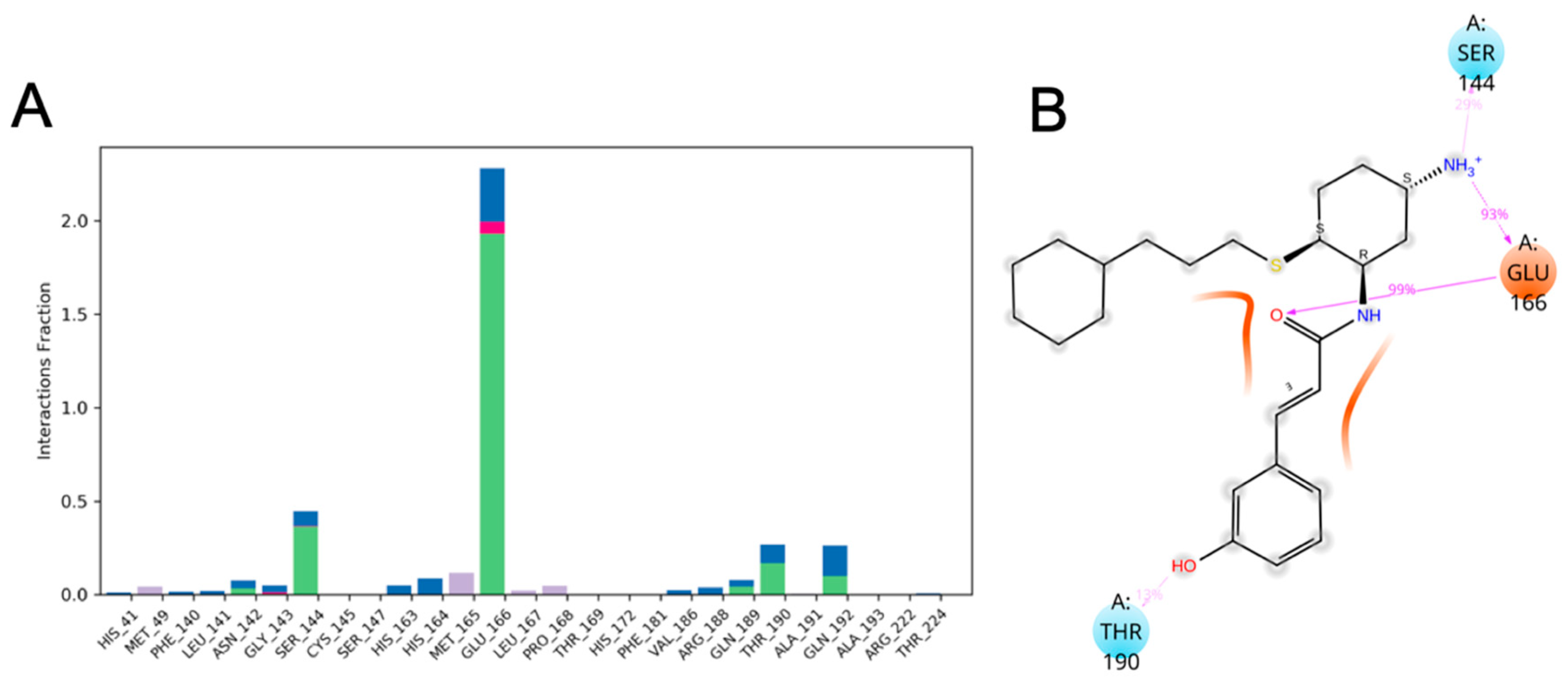
3.10. Molecular Dynamics of CM02, CM06, and CM07 for SARS-CoV-2 Mpro (PDB ID: 6LU7)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ksiazek, T.G.; Erdman, D.; Goldsmith, C.S.; Zaki, S.R.; Peret, T.; Emery, S.; Tong, S.; Urbani, C.; Comer, J.A.; Lim, W.; et al. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1953–1966. [Google Scholar] [CrossRef] [PubMed]
- Drosten, C.; Günther, S.; Preiser, W.; Van Der Werf, S.; Brodt, H.R.; Becker, S.; Rabenau, H.; Panning, M.; Kolesnikova, L.; Fouchier, R.A.; et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N. Engl. J. Med. 2003, 348, 1967–1976. [Google Scholar] [CrossRef]
- Kuiken, T.; Fouchier, R.A.; Schutten, M.; Rimmelzwaan, G.F.; Van Amerongen, G.; Van Riel, D.; Laman, J.D.; De Jong, T.; Van Doornum, G.; Lim, W.; et al. Newly discovered coronavirus as the primary cause of severe acute respiratory syndrome. Lancet 2003, 362, 263–270. [Google Scholar] [CrossRef]
- Zaki, A.M.; Van Boheemen, S.; Bestebroer, T.M.; Osterhaus, A.D.; Fouchier, R.A. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N. Engl. J. Med. 2012, 367, 1814–1820. [Google Scholar] [CrossRef]
- Chan, J.F.; Li, K.S.; To, K.K.; Cheng, V.C.; Chen, H.; Yuen, K.Y. Is the discovery of the novel human betacoronavirus 2c EMC/2012 (HCoV-EMC) the beginning of another SARS-like pandemic? J. Infect. 2012, 65, 477–489. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A novel coronavirus from patients with pneumonia in China, 2019. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Johns Hopkins University of Medicine Coronavirus Resource Center. Available online: https://coronavirus.jhu.edu/map.html (accessed on 17 July 2020).
- Hilgenfeld, R. From SARS to MERS: Crystallographic studies on coronaviral proteases enable antiviral drug design. FEBS J. 2014, 281, 4085–4096. [Google Scholar] [CrossRef] [PubMed]
- Sterling, T.; Irwin, J.J. ZINC15 – Ligand Discovery for Everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M pro from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Dai, W.; Zhang, B.; Jiang, X.M.; Su, H.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.; Peng, J.; Liu, F.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Zhang, L.; Lin, D.; Sun, X.; Curth, U.; Drosten, C.; Sauerhering, L.; Becker, S.; Rox, K.; Hilgenfeld, R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Yang, M.; Ding, Y.; Liu, Y.; Lou, Z.; Zhou, Z.; Sun, L.; Mo, L.; Ye, S.; Pang, H.; et al. The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor. Proc. Natl. Acad. Sci. USA 2003, 100, 13190–13195. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar] [CrossRef]
- Pawar, A.Y. Combating devastating COVID-19 by drug repurposing. Int. J. Antimicrob. Ag. 2020, in press. [Google Scholar] [CrossRef] [PubMed]
- Shah, B.; Modi, P.; Sagar, S.R. In silico studies on therapeutic agents for COVID-19: Drug repurposing approach. Life Sci. 2020, 117652. [Google Scholar] [CrossRef] [PubMed]
- Khan, R.J.; Jha, R.K.; Amera, G.M.; Jain, M.; Singh, E.; Pathak, A.; Singh, R.P.; Muthukumaran, J.; Singh, A.K. Targeting SARS-CoV-2: A systematic drug repurposing approach to identify promising inhibitors against 3C-like proteinase and 2′-O-ribose methyltransferase. J. Biomol. Struct. Dyn. 2020, 1–14. [Google Scholar] [CrossRef]
- Arun, K.G.; Sharanya, C.S.; Abhithaj, J.; Francis, D.; Sadasivan, C. Drug repurposing against SARS-CoV-2 using E-pharmacophore based virtual screening, molecular docking and molecular dynamics with main protease as the target. J. Biomol. Struct. Dyn. 2020, 1–12. [Google Scholar] [CrossRef]
- Dyall, J.; Coleman, C.M.; Hart, B.J.; Venkataraman, T.; Holbrook, M.R.; Kindrachuk, J.; Johnson, R.F.; Olinger, G.G.; Jahrling, P.B.; Laidlaw, M.; et al. Repurposing of clinically developed drugs for treatment of Middle East respiratory syndrome coronavirus infection. Antimicrob Agents Chem. 2014, 58, 4885–4893. [Google Scholar] [CrossRef]
- Odhar, H.A.; Ahjel, S.W.; Albeer, A.A.M.A.; Hashim, A.F.; Rayshan, A.M.; Humadi, S.S. Molecular docking and dynamics simulation of FDA approved drugs with the main protease from 2019 novel coronavirus. Bioinformation 2020, 16, 236. [Google Scholar] [CrossRef]
- Yu, R.; Chen, L.; Lan, R.; Shen, R.; Li, P. Computational screening of antagonist against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int. J. Antimicrob. Ag. 2020, 106012. [Google Scholar] [CrossRef]
- Miceli, L.A.; Teixeira, V.L.; Castro, H.C.; Rodrigues, C.R.; Mello, J.F.; Albuquerque, M.G.; Cabral, L.M.; De Brito, M.A.; De Souza, A.M. Molecular docking studies of marine diterpenes as inhibitors of wild-type and mutants HIV-1 reverse transcriptase. Mar. Drugs 2013, 11, 4127–4143. [Google Scholar] [CrossRef] [PubMed]
- Liu, G.; Wang, W.; Wan, Y.; Ju, X.; Gu, S. Application of 3D-QSAR, pharmacophore, and molecular docking in the molecular design of diarylpyrimidine derivatives as HIV-1 nonnucleoside reverse transcriptase inhibitors. Int. J. Mol. Sci. 2018, 19, 1436. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.M.; Patel, L.J. Design, synthesis, molecular docking, and antibacterial evaluation of some novel flouroquinolone derivatives as potent antibacterial agent. Sci. World J. 2014. [Google Scholar] [CrossRef] [PubMed]
- Gullapelli, K.; Brahmeshwari, G.; Ravichander, M.; Kusuma, U. Synthesis, antibacterial and molecular docking studies of new benzimidazole derivatives. Egypt. J. Basic Appl. Sci. 2017, 4, 303–309. [Google Scholar] [CrossRef]
- Stoddard, S.V.; May, X.A.; Rivas, F.; Dodson, K.; Vijayan, S.; Adhika, S.; Parker, K.; Watkins, D.L. Design of Potent Panobinostat Histone Deacetylase Inhibitor Derivatives: Molecular Considerations for Enhanced Isozyme Selectivity between HDAC2 and HDAC8. Mol. Inform. 2019, 38, 1800080. [Google Scholar] [CrossRef]
- Balasubramaniam, S.; Vijayan, S.; Goldman, L.V.; May, X.A.; Dodson, K.; Adhikari, S.; Rivas, F.; Watkins, D.L.; Stoddard, S.V. Design and synthesis of diazine-based panobinostat analogues for HDAC8 inhibition. Beilstein J. Org. Chem. 2020, 16, 628–637. [Google Scholar] [CrossRef]
- Stoddard, S.V.; Dodson, K.; Adams, K.; Watkins, D.L. In silico Design of Novel Histone Deacetylase 4 Inhibitors: Design Guidelines for Improved Binding Affinity. Int. J. Mol. Sci. 2020, 21, 219. [Google Scholar] [CrossRef]
- Fearon, D.; Powell, A.J.; Douanamath, A.; Owen, C.D.; Wild, C.; Krojer, T.; Lukacik, P.; Strain-Damerell, C.M.; Walsh, M.A.; von Delft, F. PanDDA analysis group deposition—Crystal Structure of SARS-CoV-2 main protease in complex with Z1220452176. 2020, unpublished. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera a visualization system for exploratory research and analysis. J. Comput. Chem. 2014, 13, 1605–1612. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex: Fully Automatic Flexible Molecular Docking Usig a Molecular Similarity-Based Search Engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef]
- Jain, A.N. Surflex-Dock 2.1: Robust performance from ligan energetic modeling, rig flexibility, and knowledge-based search. J. Comput. Aid. Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Glaser, F.; Pupko, T.; Paz, I.; Bell, R.E.; Bechor-Shental, D.; Martz, E.; Ben-Tal, N. ConSurf: Identification of functional regions in proteins by surface-mapping of phylogenetic information. Bioinformatics 2003, 19, 163–164. [Google Scholar] [CrossRef] [PubMed]
- Landau, M.; Mayrose, I.; Rosenberg, Y.; Glaser, F.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2005: The projection of evolutionary conservation scores of residues on protein structures. Nucleic Acids Res. 2005, 33, W299–W302. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V.; Gasteiger, J.; Todeschini, R.; Mauri, A.; Livingstone, D.; Ertl, P.; Palyulin, V.A.; Radchenko, E.V.; Zefirov, N.S.; Makarenko, A.S.; et al. Virtual computational chemistry laboratory—Design and description. J. Comput. Aid. Mol. Des. 2005, 19, 453–463. [Google Scholar] [CrossRef]
- Molinspiration Cheminformatics Free Web Services; Molecular Property and Bioactivity Score Calculation Toolkit. Available online: https://www.molinspiration.com (accessed on 18 July 2020).
- Ertl, P.; Rohde, B.; Selzer, P. Fast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- D.E. Shaw Research. Shaw Research Desmond Molecular Dynamics System; Maestro-Desmond Interoperability Tools; Schrödinger, LLC: New York, NY, USA, 2019. [Google Scholar]
- Jorgensen, W.L. Quantum and Statistical Mechanical Studies of Liquids: 10. Transferable Intermolecular Potential Functions for Water, Alcohols, and Ethers. Application to Liquid Water. J. Am. Chem. Soc. 1981, 103, 335–340. [Google Scholar] [CrossRef]
- Chaurasiya, N.D.; Zhao, J.; Pandey, P.; Doerksen, R.J.; Muhammad, I.; Tekwani, B.L. Selective Inhibition of Human Monoamine Oxidase B by Acacetin 7-Methyl Ether Isolated from Turnera diffusa (Damiana). Molecules 2019, 24, 810. [Google Scholar] [CrossRef]
- Huber, R.G.; Margreiter, M.A.; Fuchs, J.E.; von Grafenstein, S.; Tautermann, C.S.; Liedl, K.R.; Fox, T. Heteroaromatic π-stacking energy landscapes. J. Chem. Inf. Model. 2014, 54, 1371–1379. [Google Scholar] [CrossRef]























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
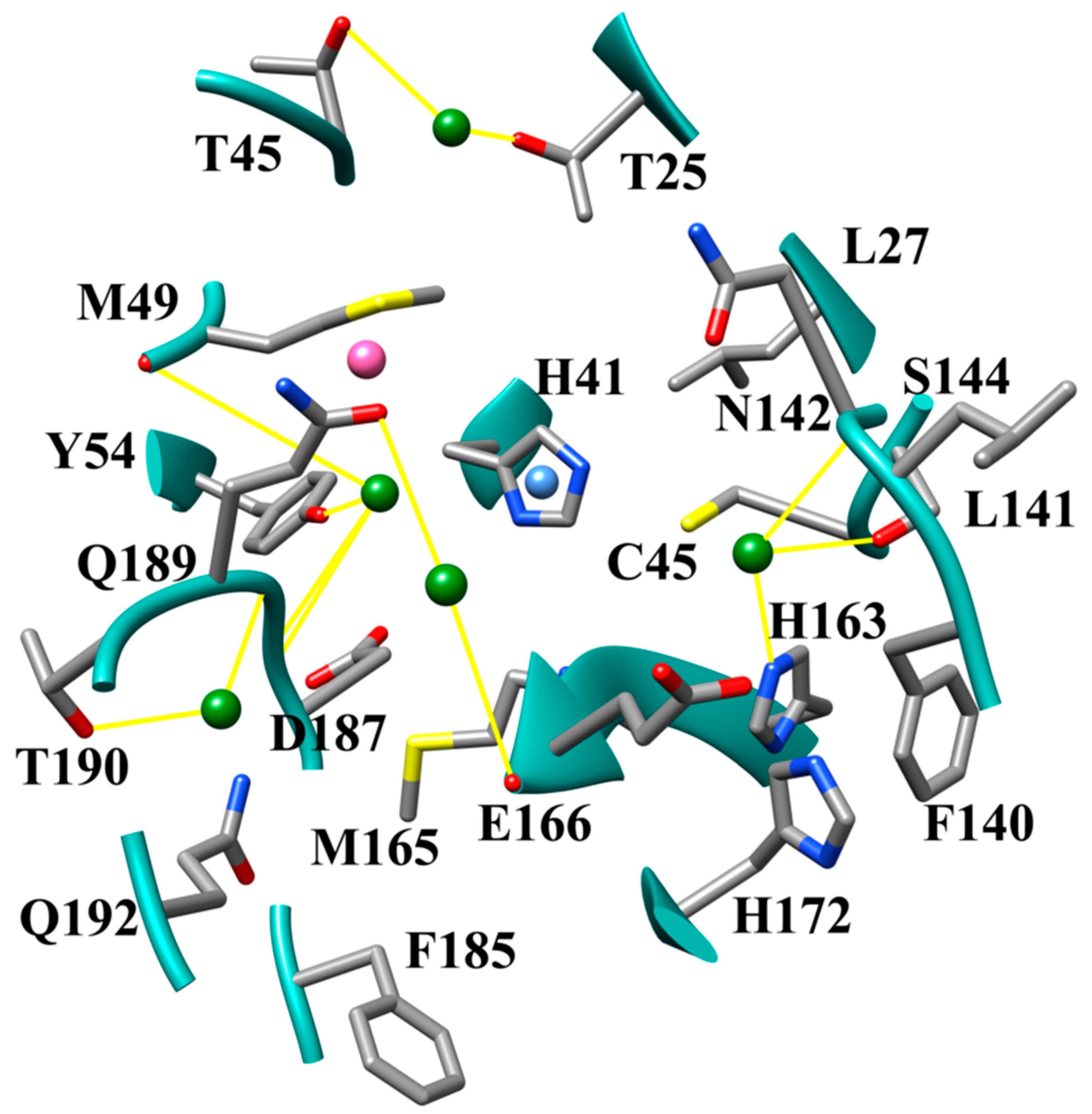{kind=link}
| Residue Position | Subsite | Residue in SARS-CoV-2 Mpro | Residue Variety |
|---|---|---|---|
| 49 | S2 | Met | Leu, Ala, Met, Tyr, Ser, Phe, Asn, Thr |
| 50 | S2 | Leu | Val, Thr, Asn, Gln, Lys, His, Ser, Arg, Met, Ala, Leu |
| 190 | S4 | Thr | Thr, Val, Lys, Asn, Cys, Ile, Arg, Ser, Leu |
| 191 | S4 | Ala | Val, Ile, Pro, Phe, His, Tyr, Ser, Met, Leu, Ala |
| 193 | S4 | Val | Lys, Asn, Gln, Ile, Val, Ala, Leu, Met, Phe, Arg, His, Ser |
| Zinc Database Inhibitor | PDB File of Compound | Abbreviated Compound Name in PDB File | Docking Score −log10(Kd) |
|---|---|---|---|
| Z1587220559 | 5REC | T1J | 5.92 |
| Z271004858 | 5RF8 | SFY | 5.42 |
| PCM-0102269 | 5RET | T47 | 4.69 |
| Z1271660837 | 5RFB | K3S | 3.80 |
| PCM-0102575 | 5RFK | T7D | 3.47 |
| Compound | Highest Docking Score −log10(Kd) |
|---|---|
| T1J G-series | 5.92 |
| GM03 | 5.64 |
| GN01 | 5.43 |
| GN02 | 5.39 |
| GM10 | 4.82 |
| T47 chair B-series | 4.69 |
| BM26 | 4.68 |
| BD11 | 4.66 |
| BD23 | 4.43 |
| BM24 | 4.17 |
| BD22 | 4.25 |
| BM25 | 4.08 |
| Compound | Highest Docking Score −log10(Kd) | Substituent Added | Compound | Highest Docking Score −log10(Kd) | o, m, p | Substituent Added |
|---|---|---|---|---|---|---|
| HS04 | 6.30 | Butyl | SD12 | 7.62 | m | Butyl |
| BD03 | 5.92 | SD11 | 7.28 | m | Propyl | |
| HS03 | 5.68 | Propyl | SD23 | 7.17 | m | Neopentyl |
| BD02 | 5.41 | SD15 | 7.16 | p | Propyl | |
| BD01 | 5.40 | SD27 | 7.04 | m | Butenyl | |
| HS01 | 5.05 | Methyl | SD28 | 6.79 | p | Butenyl |
| HS02 | 4.95 | Ethyl | SD07 | 6.66 | o | Propyl |
| T47 | 4.69 | SD08 | 6.53 | o | Butyl | |
| SD24 | 6.45 | p | Neopentyl | |||
| SD16 | 6.35 | p | Butyl | |||
| SD14 | 6.20 | p | Ethyl | |||
| SD26 | 6.10 | o | Butenyl | |||
| SD09 | 6.10 | m | Methyl | |||
| SD22 | 6.07 | o | Neopentyl | |||
| SD10 | 5.95 | m | Ethyl | |||
| SD13 | 5.88 | p | Methyl | |||
| SD06 | 5.70 | o | Ethyl | |||
| SD05 | 5.51 | o | Methyl | |||
| SFY | 5.42 |
| Compound | Highest Docking Score −log10(Kd) | Compound | Highest Docking Score −log10(Kd) |
|---|---|---|---|
| DB04 | 6.88 | SD29 | 7.72 |
| DB09 | 6.76 | SD25 | 7.19 |
| DB11 | 6.77 | SD19 | 6.83 |
| DB03 | 6.65 | SD31 | 6.68 |
| DB12 | 6.33 | SD18 | 6.21 |
| DB02 | 6.32 | SD20 | 5.46 |
| DB10 | 6.22 | SFY | 5.42 |
| DB08 | 5.97 | SD30 | 5.42 |
| DB05 | 5.53 | SD17 | 5.40 |
| DB06 | 5.39 | SD32 | 4.69 |
| T47 | 4.69 | SD33 | 4.31 |
| Compound | Highest Docking Score −log10(Kd) | OMP | Substituent(s) | Compound | Highest Docking Score −log10(Kd) |
|---|---|---|---|---|---|
| SD34 | 11.02 | M/P | Butyl/butyl | KT11 | 7.00 |
| LEA4 | 10.94 | O/P | Butyl/butyl | E010 | 6.97 |
| LEA2 | 10.02 | O/P | Propyl/propyl | KT10 | 6.36 |
| KT04 | 9.92 | P | Butyl | KT14 | 6.09 |
| KTH1 | 9.82 | O/P | Ethyl/butyl | KT12 | 5.72 |
| KTH3 | 9.82 | Butyl/cyclohexyl | EW10 | 5.52 | |
| SD35 | 9.72 | M/P | Propyl/propyl | EW01 | 4.36 |
| KTH2 | 9.64 | O/P | Methyl/butyl | EW06 | 4.27 |
| LEA3 | 9.54 | O/P | Ethyl/ethyl | EW08 | 4.17 |
| SD04 | 9.41 | M | Butyl | K3S | 3.80 |
| SD03 | 9.33 | M | Propyl | EW07 | 3.69 |
| SD36 | 9.21 | M/P | Ethyl/ethyl | ||
| KT03 | 9.21 | P | Propyl | ||
| LM03 | 9.12 | O | Butyl | ||
| LEA1 | 8.81 | O/P | Methyl/methyl | ||
| L006 | 8.75 | O | Propyl | ||
| KT02 | 8.71 | P | Ethyl | ||
| SD37 | 8.62 | M/P | Methyl/methyl | ||
| SD02 | 8.52 | M | Ethyl | ||
| L005 | 8.10 | O | Ethyl | ||
| L001 | 8.26 | ||||
| LM02 | 8.28 | ||||
| KT01 | 7.91 | P | Methyl | ||
| SD01 | 7.83 | M | Methyl | ||
| SD21 | 7.74 | ||||
| L004 | 7.60 | O | Methyl | ||
| SFY | 5.42 |
| Compound | Highest Docking Score −log10(Kd) | Compound | Highest Docking Score −log10(Kd) |
|---|---|---|---|
| KF08 | 8.80 | JN16 | 8.16 |
| KF03 | 8.07 | JMH5 | 7.42 |
| KF04 | 7.14 | JN14 | 6.00 |
| KF07 | 6.90 | JCN6 | 5.82 |
| KF01 | 6.77 | JCN8 | 5.79 |
| KF02 | 6.66 | JN12 | 5.61 |
| GN06 | 6.41 | JN10 | 5.28 |
| T1J | 5.92 | T7DM | 3.77 |
| GN07 | 5.67 | T7D | 3.47 |
| GM09 | 5.61 | ||
| KF05 | 5.52 | ||
| EW13 | 7.33 | HS06 | 7.85 |
| EW14 | 7.32 | HS05 | 7.03 |
| EW11 | 6.99 | T47 | 4.69 |
| Compound | Highest Docking Score −log10(Kd) | LogP | LogS |
|---|---|---|---|
| FL30 | 13.98 | 1.62 | −4.27 |
| FL20 | 13.17 | 1.44 | −3.76 |
| FL29 | 12.98 | 4.39 | −5.51 |
| FL28 | 12.98 | 1.62 | −4.01 |
| FL23 | 12.96 | 4.25 | −5.38 |
| FL26 | 12.67 | 4.35 | −5.56 |
| FL16 | 12.41 | 0.79 | −3.23 |
| FL22 | 12.32 | 1.43 | −3.72 |
| FL24 | 12.30 | 4.23 | −5.32 |
| KBH1 | 11.31 | 1.96 | −5.48 |
| FL05 | 11.11 | 0.45 | −2.67 |
| FL18 | 11.09 | 1.00 | −3.56 |
| FL14 | 10.83 | 1.15 | −3.65 |
| FL09 | 10.62 | 0.63 | −2.82 |
| FL04 | 10.60 | 0.45 | −2.64 |
| FL21 | 10.50 | 2.00 | −3.58 |
| KB01 | 10.46 | 0.90 | −4.46 |
| FL27 | 10.44 | 0.58 | −2.87 |
| FL31 | 10.27 | 1.39 | −3.79 |
| FL15 | 10.10 | 1.13 | −3.65 |
| FL06 | 10.05 | 0.86 | −2.91 |
| FL08 | 9.92 | 0.11 | −2.38 |
| FL07 | 9.82 | 0.14 | −2.38 |
| FL25 | 9.42 | 0.29 | −2.84 |
| FL03 | 9.26 | 1.26 | −3.36 |
| T1J | 5.92 | 2.83 | −3.16 |
| SFY | 5.42 | 0.84 | −3.03 |
| T47 | 4.69 | 1.73 | −2.02 |
| K3S | 3.80 | −0.27 | −1.43 |
| T7D | 3.47 | 1.73 | −2.02 |
| Inhibitor Name | Highest Docking Score −log10(Kd) | LogP | LogS | MW | Topological Polar Surface Area | Protease Bioactivity Score |
|---|---|---|---|---|---|---|
| CM06 | 10.60 | 3.61 | −5.74 | 402.61 | 80.91 | 0.51 |
| CM07 | 9.12 | 3.75 | −6.03 | 410.59 | 93.17 | 0.11 |
| CM02 | 8.98 | 5.23 | −6.12 | 416.63 | 75.35 | 0.46 |
| CO08 | 8.95 | 5.51 | −6.54 | 404.63 | 55.12 | 0.49 |
| CM05 | 8.42 | 4.39 | −6.13 | 417.25 | 58.36 | 0.56 |
| CM01 | 8.29 | 2.66 | −4.58 | 407.67 | 58.36 | 0.76 |
| CO09 | 8.19 | 5.51 | −6.55 | 400.63 | 55.12 | 0.47 |
| CO03 | 8.15 | 3.41 | −5.06 | 367.60 | 58.36 | 0.60 |
| CO04 | 8.05 | 2.53 | −4.85 | 361.56 | 58.36 | 0.46 |
| CO10 | 7.28 | 5.84 | −6.63 | 394.58 | 55.12 | 0.21 |
| Cinanserin | 6.01 | 4.27 | −5.24 | 340.49 | 32.34 | −0.10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoddard, S.V.; Stoddard, S.D.; Oelkers, B.K.; Fitts, K.; Whalum, K.; Whalum, K.; Hemphill, A.D.; Manikonda, J.; Martinez, L.M.; Riley, E.G.; et al. Optimization Rules for SARS-CoV-2 Mpro Antivirals: Ensemble Docking and Exploration of the Coronavirus Protease Active Site. Viruses 2020, 12, 942. https://doi.org/10.3390/v12090942
Stoddard SV, Stoddard SD, Oelkers BK, Fitts K, Whalum K, Whalum K, Hemphill AD, Manikonda J, Martinez LM, Riley EG, et al. Optimization Rules for SARS-CoV-2 Mpro Antivirals: Ensemble Docking and Exploration of the Coronavirus Protease Active Site. Viruses. 2020; 12(9):942. https://doi.org/10.3390/v12090942
Chicago/Turabian StyleStoddard, Shana V., Serena D. Stoddard, Benjamin K. Oelkers, Kennedi Fitts, Kellen Whalum, Kaylah Whalum, Alexander D. Hemphill, Jithin Manikonda, Linda Michelle Martinez, Elizabeth G. Riley, and et al. 2020. "Optimization Rules for SARS-CoV-2 Mpro Antivirals: Ensemble Docking and Exploration of the Coronavirus Protease Active Site" Viruses 12, no. 9: 942. https://doi.org/10.3390/v12090942
APA StyleStoddard, S. V., Stoddard, S. D., Oelkers, B. K., Fitts, K., Whalum, K., Whalum, K., Hemphill, A. D., Manikonda, J., Martinez, L. M., Riley, E. G., Roof, C. M., Sarwar, N., Thomas, D. M., Ulmer, E., Wallace, F. E., Pandey, P., & Roy, S. (2020). Optimization Rules for SARS-CoV-2 Mpro Antivirals: Ensemble Docking and Exploration of the Coronavirus Protease Active Site. Viruses, 12(9), 942. https://doi.org/10.3390/v12090942