Cell Cycle Regulation in Macrophages and Susceptibility to HIV-1


{kind=link}
{kind=link}
Abstract
1. Introduction
2. Macrophage Origin and Polarization/Activation
2.1. Origin
2.2. Localization
3. Macrophages and HIV Infection
4. Cell Cycle Regulation in Macrophages
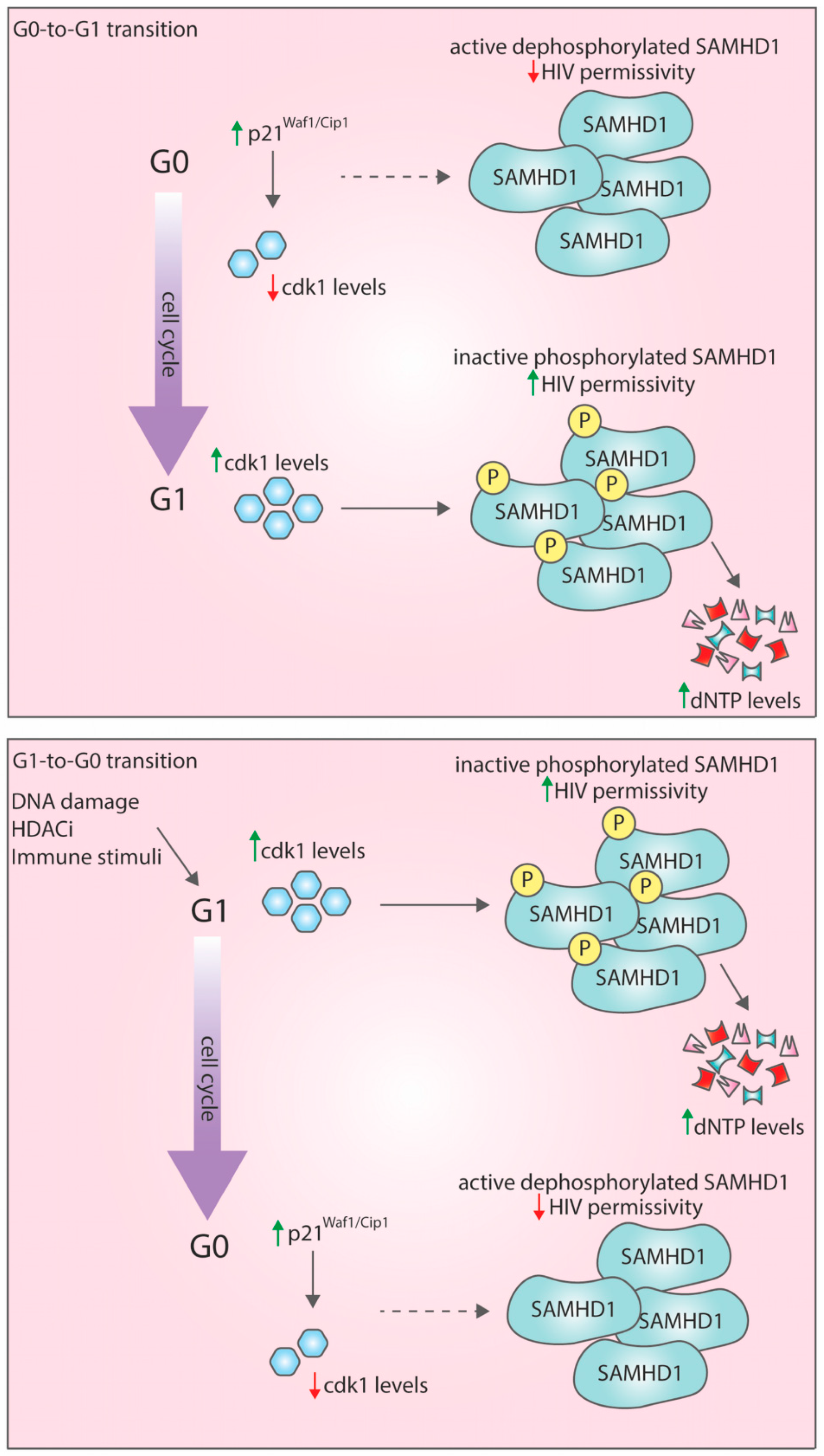
4.1. G0-to-G1 Transition: A Window of Opportunity for HIV-1 Infection
4.2. G1-to-G0 Transition
4.3. Histone Deacetylase Inhibitors (HDACi)
4.4. DNA Damage
4.5. Immune Stimuli and Gram-Negative Bacteria
5. HIV-1 Regulates the Host Cell Cycle
5.1. Viral Protein R (Vpr)
5.2. Viral Infectivity Factor, Vif
5.3. What Is the Role of Vpr/Vif-Mediated G2/M Arrest in Terminally Differentiated Cells Such as Macrophages?
6. Conclusions
Funding
Conflicts of Interest
References
- Churchill, M.J.; Deeks, S.G.; Margolis, D.M.; Siliciano, R.F.; Swanstrom, R. Hiv reservoirs: What, where and how to target them. Nat. Rev. Microbiol. 2015, 14, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Eisele, E.; Siliciano, R.F. Redefining the viral reservoirs that prevent hiv-1 eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef] [PubMed]
- Volberding, P.A.; Deeks, S.G. Antiretroviral therapy and management of hiv infection. Lancet 2010, 376, 49–62. [Google Scholar] [CrossRef]
- Autran, B.; Descours, B.; Bacchus, C. Immune control of hiv-1 reservoirs. Curr. Opin. HIV AIDS 2013, 8, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.; Zhang, H.; Lopez, P.; Pardo, C.A.; Gartner, S. In vitro modeling of the hiv-macrophage reservoir. J. Leukoc. Biol. 2006, 80, 1127–1135. [Google Scholar] [CrossRef]
- Castellano, P.; Prevedel, L.; Eugenin, E.A. Hiv-infected macrophages and microglia that survive acute infection become viral reservoirs by a mechanism involving bim. Sci. Rep. 2017, 7, 12866. [Google Scholar] [CrossRef]
- Abbas, W.; Tariq, M.; Iqbal, M.; Kumar, A.; Herbein, G. Eradication of hiv-1 from the macrophage reservoir: An uncertain goal? Viruses 2015, 7, 1578–1598. [Google Scholar] [CrossRef]
- Honeycutt, J.B.; Thayer, W.O.; Baker, C.E.; Ribeiro, R.M.; Lada, S.M.; Cao, Y.; Cleary, R.A.; Hudgens, M.G.; Richman, D.D.; Garcia, J.V. Hiv persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nat. Med. 2017, 23, 638–643. [Google Scholar] [CrossRef]
- Ganor, Y.; Real, F.; Sennepin, A.; Dutertre, C.A.; Prevedel, L.; Xu, L.; Tudor, D.; Charmeteau, B.; Couedel-Courteille, A.; Marion, S.; et al. Hiv-1 reservoirs in urethral macrophages of patients under suppressive antiretroviral therapy. Nat. Microbiol. 2019, 4, 633–644. [Google Scholar] [CrossRef]
- Pelchen-Matthews, A.; Kramer, B.; Marsh, M. Infectious hiv-1 assembles in late endosomes in primary macrophages. J. Cell Biol. 2003, 162, 443–455. [Google Scholar] [CrossRef]
- Gupta, R.K.; Abdul-Jawad, S.; McCoy, L.E.; Mok, H.P.; Peppa, D.; Salgado, M.; Martinez-Picado, J.; Nijhuis, M.; Wensing, A.M.J.; Lee, H.; et al. Hiv-1 remission following ccr5delta32/delta32 haematopoietic stem-cell transplantation. Nature 2019, 568, 244–248. [Google Scholar] [CrossRef] [PubMed]
- Gupta, R.K.; Peppa, D.; Hill, A.L.; Gálvez, C.; Salgado, M.; Pace, M.; McCoy, L.E.; Griffith, S.A.; Thornhill, J.; Alrubayyi, A.; et al. Evidence for hiv-1 cure after ccr5δ32/δ32 allogeneic haemopoietic stem-cell transplantation 30 months post analytical treatment interruption: A case report. Lancet HIV 2020. [Google Scholar] [CrossRef]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müßig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-term control of hiv by ccr5 delta32/delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Gentek, R.; Molawi, K.; Sieweke, M.H. Tissue macrophage identity and self-renewal. Immunol. Rev. 2014, 262, 56–73. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.E.; Jaworowski, A.; Hearps, A.C. The hiv reservoir in monocytes and macrophages. Front. Immunol. 2019, 10, 1435. [Google Scholar] [CrossRef] [PubMed]
- Fois, A.F.; Brew, B.J. The potential of the cns as a reservoir for hiv-1 infection: Implications for hiv eradication. Curr. HIV/AIDS Rep. 2015, 12, 299–303. [Google Scholar] [CrossRef]
- Hellmuth, J.; Valcour, V.; Spudich, S. Cns reservoirs for hiv: Implications for eradication. J. Virus Erad. 2015, 1, 67–71. [Google Scholar] [CrossRef]
- Langford, D.; Marquie-Beck, J.; de Almeida, S.; Lazzaretto, D.; Letendre, S.; Grant, I.; McCutchan, J.A.; Masliah, E.; Ellis, R.J. Relationship of antiretroviral treatment to postmortem brain tissue viral load in human immunodeficiency virus-infected patients. J. Neurovirol. 2006, 12, 100–107. [Google Scholar] [CrossRef]
- Smith, D.B.; Simmonds, P.; Bell, J.E. Brain viral burden, neuroinflammation and neurodegeneration in haart-treated hiv positive injecting drug users. J. Neurovirol. 2014, 20, 28–38. [Google Scholar] [CrossRef]
- Akiyama, H.; Gummuluru, S. Hiv-1 persistence and chronic induction of innate immune responses in macrophages. Viruses 2020, 12, 711. [Google Scholar] [CrossRef]
- Aquaro, S.; Borrajo, A.; Pellegrino, M.; Svicher, V. Mechanisms underlying of antiretroviral drugs in different cellular reservoirs with a focus on macrophages. Virulence 2020, 11, 400–413. [Google Scholar] [CrossRef] [PubMed]
- Boliar, S.; Gludish, D.W.; Jambo, K.C.; Kamng’ona, R.; Mvaya, L.; Mwandumba, H.C.; Russell, D.G. Inhibition of the lncrna saf drives activation of apoptotic effector caspases in hiv-1-infected human macrophages. Proc. Natl. Acad. Sci. USA 2019, 116, 7431–7438. [Google Scholar] [CrossRef] [PubMed]
- Swingler, S.; Mann, A.M.; Zhou, J.; Swingler, C.; Stevenson, M. Apoptotic killing of hiv-1-infected macrophages is subverted by the viral envelope glycoprotein. PLoS Pathog. 2007, 3, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A.; Vannella, K.M. Macrophages in tissue repair, regeneration, and fibrosis. Immunity 2016, 44, 450–462. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Lavine, K.J.; Randolph, G.J. Origin and functions of tissue macrophages. Immunity 2014, 41, 21–35. [Google Scholar] [CrossRef] [PubMed]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef]
- Hashimoto, D.; Chow, A.; Noizat, C.; Teo, P.; Beasley, M.B.; Leboeuf, M.; Becker, C.D.; See, P.; Price, J.; Lucas, D.; et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity 2013, 38, 792–804. [Google Scholar] [CrossRef]
- Yona, S.; Kim, K.W.; Wolf, Y.; Mildner, A.; Varol, D.; Breker, M.; Strauss-Ayali, D.; Viukov, S.; Guilliams, M.; Misharin, A.; et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 2013, 38, 79–91. [Google Scholar] [CrossRef]
- Nayak, D.K.; Zhou, F.; Xu, M.; Huang, J.; Tsuji, M.; Hachem, R.; Mohanakumar, T. Long-term persistence of donor alveolar macrophages in human lung transplant recipients that influences donor-specific immune responses. Am. J. Transplant. 2016, 16, 2300–2311. [Google Scholar] [CrossRef]
- Guilliams, M.; De Kleer, I.; Henri, S.; Post, S.; Vanhoutte, L.; De Prijck, S.; Deswarte, K.; Malissen, B.; Hammad, H.; Lambrecht, B.N. Alveolar macrophages develop from fetal monocytes that differentiate into long-lived cells in the first week of life via gm-csf. J. Exp. Med. 2013, 210, 1977–1992. [Google Scholar] [CrossRef]
- Zaslona, Z.; Przybranowski, S.; Wilke, C.; van Rooijen, N.; Teitz-Tennenbaum, S.; Osterholzer, J.J.; Wilkinson, J.E.; Moore, B.B.; Peters-Golden, M. Resident alveolar macrophages suppress, whereas recruited monocytes promote, allergic lung inflammation in murine models of asthma. J. Immunol. 2014, 193, 4245–4253. [Google Scholar] [CrossRef] [PubMed]
- Byrne, A.J.; Powell, J.E.; O’Sullivan, B.J.; Ogger, P.P.; Hoffland, A.; Cook, J.; Bonner, K.L.; Hewitt, R.J.; Wolf, S.; Ghai, P.; et al. Dynamics of human monocytes and airway macrophages during healthy aging and after transplant. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Bajpai, G.; Schneider, C.; Wong, N.; Bredemeyer, A.; Hulsmans, M.; Nahrendorf, M.; Epelman, S.; Kreisel, D.; Liu, Y.; Itoh, A.; et al. The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 2018, 24, 1234–1245. [Google Scholar] [CrossRef] [PubMed]
- Orecchioni, M.; Ghosheh, Y.; Pramod, A.B.; Ley, K. Macrophage polarization: Different gene signatures in m1(lps+) vs. Classically and m2(lps-) vs. Alternatively activated macrophages. Front. Immunol. 2019, 10, 1084. [Google Scholar] [CrossRef]
- Gosselin, D.; Skola, D.; Coufal, N.G.; Holtman, I.R.; Schlachetzki, J.C.M.; Sajti, E.; Jaeger, B.N.; O’Connor, C.; Fitzpatrick, C.; Pasillas, M.P.; et al. An environment-dependent transcriptional network specifies human microglia identity. Science 2017, 356. [Google Scholar] [CrossRef]
- Lavin, Y.; Kobayashi, S.; Leader, A.; Amir, E.D.; Elefant, N.; Bigenwald, C.; Remark, R.; Sweeney, R.; Becker, C.D.; Levine, J.H.; et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 2017, 169, 750–765.E17. [Google Scholar] [CrossRef]
- Xue, J.; Schmidt, S.V.; Sander, J.; Draffehn, A.; Krebs, W.; Quester, I.; De Nardo, D.; Gohel, T.D.; Emde, M.; Schmidleithner, L.; et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity 2014, 40, 274–288. [Google Scholar] [CrossRef]
- Cassetta, L.; Pollard, J.W. Targeting macrophages: Therapeutic approaches in cancer. Nat. Rev. Drug Discov. 2018, 17, 887–904. [Google Scholar] [CrossRef]
- Vitale, I.; Manic, G.; Coussens, L.M.; Kroemer, G.; Galluzzi, L. Macrophages and metabolism in the tumor microenvironment. Cell Metab. 2019, 30, 36–50. [Google Scholar] [CrossRef]
- Mosser, D.M.; Edwards, J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008, 8, 958. [Google Scholar] [CrossRef]
- Cribbs, S.K.; Lennox, J.; Caliendo, A.M.; Brown, L.A.; Guidot, D.M. Healthy hiv-1-infected individuals on highly active antiretroviral therapy harbor hiv-1 in their alveolar macrophages. AIDS Res. Hum. Retrovir. 2015, 31, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Deleage, C.; Chan, C.N.; Busman-Sahay, K.; Estes, J.D. Next-generation in situ hybridization approaches to define and quantify hiv and siv reservoirs in tissue microenvironments. Retrovirology 2018, 15, 4. [Google Scholar] [CrossRef] [PubMed]
- DiNapoli, S.R.; Ortiz, A.M.; Wu, F.; Matsuda, K.; Twigg, H.L., 3rd; Hirsch, V.M.; Knox, K.; Brenchley, J.M. Tissue-resident macrophages can contain replication-competent virus in antiretroviral-naive, siv-infected asian macaques. JCI Insight 2017, 2, e91214. [Google Scholar] [CrossRef] [PubMed]
- Jambo, K.C.; Banda, D.H.; Kankwatira, A.M.; Sukumar, N.; Allain, T.J.; Heyderman, R.S.; Russell, D.G.; Mwandumba, H.C. Small alveolar macrophages are infected preferentially by hiv and exhibit impaired phagocytic function. Mucosal. Immunol. 2014, 7, 1116–1126. [Google Scholar] [CrossRef]
- Kandathil, A.J.; Sugawara, S.; Goyal, A.; Durand, C.M.; Quinn, J.; Sachithanandham, J.; Cameron, A.M.; Bailey, J.R.; Perelson, A.S.; Balagopal, A. No recovery of replication-competent hiv-1 from human liver macrophages. J. Clin. Investig. 2018, 128, 4501–4509. [Google Scholar] [CrossRef]
- Tso, F.Y.; Kang, G.; Kwon, E.H.; Julius, P.; Li, Q.; West, J.T.; Wood, C. Brain is a potential sanctuary for subtype c hiv-1 irrespective of art treatment outcome. PLoS ONE 2018, 13, e0201325. [Google Scholar] [CrossRef]
- Zalar, A.; Figueroa, M.I.; Ruibal-Ares, B.; Bare, P.; Cahn, P.; de Bracco, M.M.; Belmonte, L. Macrophage hiv-1 infection in duodenal tissue of patients on long term haart. Antiviral. Res. 2010, 87, 269–271. [Google Scholar] [CrossRef]
- Li, L.; Meng, G.; Graham, M.F.; Shaw, G.M.; Smith, P.D. Intestinal macrophages display reduced permissiveness to human immunodeficiency virus 1 and decreased surface ccr5. Gastroenterology 1999, 116, 1043–1053. [Google Scholar] [CrossRef]
- Shen, R.; Richter, H.E.; Clements, R.H.; Novak, L.; Huff, K.; Bimczok, D.; Sankaran-Walters, S.; Dandekar, S.; Clapham, P.R.; Smythies, L.E.; et al. Macrophages in vaginal but not intestinal mucosa are monocyte-like and permissive to human immunodeficiency virus type 1 infection. J. Virol. 2009, 83, 3258–3267. [Google Scholar] [CrossRef]
- Goldstone, D.C.; Ennis-Adeniran, V.; Hedden, J.J.; Groom, H.C.T.; Rice, G.I.; Christodoulou, E.; Walker, P.A.; Kelly, G.; Haire, L.F.; Yap, M.W.; et al. Hiv-1 restriction factor samhd1 is a deoxynucleoside triphosphate triphosphohydrolase. Nature 2011, 480, 379–382. [Google Scholar] [CrossRef]
- Franzolin, E.; Pontarin, G.; Rampazzo, C.; Miazzi, C.; Ferraro, P.; Palumbo, E.; Reichard, P.; Bianchi, V. The deoxynucleotide triphosphohydrolase samhd1 is a major regulator of DNA precursor pools in mammalian cells. Proc. Natl. Acad. Sci. USA 2013, 110, 14272–14277. [Google Scholar] [CrossRef] [PubMed]
- Daddacha, W.; Koyen, A.E.; Bastien, A.J.; Head, P.E.; Dhere, V.R.; Nabeta, G.N.; Connolly, E.C.; Werner, E.; Madden, M.Z.; Daly, M.B.; et al. Samhd1 promotes DNA end resection to facilitate DNA repair by homologous recombination. Cell Rep. 2017, 20, 1921–1935. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, J.M.; Gelais, C.S.; de Silva, S.; Yount, J.S.; Tang, C.; Ji, X.; Shepard, C.; Xiong, Y.; Kim, B.; Wu, L. Samhd1-mediated hiv-1 restriction in cells does not involve ribonuclease activity. Nat. Med. 2016, 22, 1072–1074. [Google Scholar] [CrossRef]
- Lahouassa, H.; Daddacha, W.; Hofmann, H.; Ayinde, D.; Logue, E.C.; Dragin, L.; Bloch, N.; Maudet, C.; Bertrand, M.; Gramberg, T.; et al. Samhd1 restricts the replication of human immunodeficiency virus type 1 by depleting the intracellular pool of deoxynucleoside triphosphates. Nat. Immunol. 2012, 13, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Schenkova, K.; Adam, T.; Erikson, E.; Lehmann-Koch, J.; Sertel, S.; Verhasselt, B.; Fackler, O.T.; Lasitschka, F.; Keppler, O.T. Samhd1’s protein expression profile in humans. J. Leukoc. Biol. 2015, 98, 5–14. [Google Scholar] [CrossRef]
- Cribier, A.; Descours, B.; Valadao, A.L.; Laguette, N.; Benkirane, M. Phosphorylation of samhd1 by cyclin a2/cdk1 regulates its restriction activity toward hiv-1. Cell Rep. 2013, 3, 1036–1043. [Google Scholar] [CrossRef]
- Mlcochova, P.; Sutherland, K.A.; Watters, S.A.; Bertoli, C.; de Bruin, R.A.; Rehwinkel, J.; Neil, S.J.; Lenzi, G.M.; Kim, B.; Khwaja, A.; et al. A g1-like state allows hiv-1 to bypass samhd1 restriction in macrophages. EMBO J. 2017, 36, 604–616. [Google Scholar] [CrossRef]
- White, T.E.; Brandariz-Nuñez, A.; Valle-Casuso, J.C.; Amie, S.; Nguyen, L.A.; Kim, B.; Tuzova, M.; Diaz-Griffero, F. The retroviral restriction ability of samhd1, but not its deoxynucleotide triphosphohydrolase activity, is regulated by phosphorylation. Cell Host Microbe 2013, 17, 441–451. [Google Scholar] [CrossRef]
- Schott, K.; Fuchs, N.V.; Derua, R.; Mahboubi, B.; Schnellbacher, E.; Seifried, J.; Tondera, C.; Schmitz, H.; Shepard, C.; Brandariz-Nunez, A.; et al. Dephosphorylation of the hiv-1 restriction factor samhd1 is mediated by pp2a-b55alpha holoenzymes during mitotic exit. Nat. Commun. 2018, 9, 2227. [Google Scholar] [CrossRef]
- Hrecka, K.; Hao, C.; Gierszewska, M.; Swanson, S.K.; Kesik-Brodacka, M.; Srivastava, S.; Florens, L.; Washburn, M.P.; Skowronski, J. Vpx relieves inhibition of hiv-1 infection of macrophages mediated by the samhd1 protein. Nature 2011, 474, 658–661. [Google Scholar] [CrossRef]
- Kaushik, R.; Zhu, X.; Stranska, R.; Wu, Y.; Stevenson, M. A cellular restriction dictates the permissivity of nondividing monocytes/macrophages to lentivirus and gammaretrovirus infection. Cell Host Microbe 2009, 6, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Laguette, N.; Sobhian, B.; Casartelli, N.; Ringeard, M.; Chable-Bessia, C.; Segeral, E.; Yatim, A.; Emiliani, S.; Schwartz, O.; Benkirane, M. Samhd1 is the dendritic- and myeloid-cell-specific hiv-1 restriction factor counteracted by vpx. Nature 2011, 474, 654–657. [Google Scholar] [CrossRef] [PubMed]
- Lenzi, G.M.; Domaoal, R.A.; Kim, D.H.; Schinazi, R.F.; Kim, B. Mechanistic and kinetic differences between reverse transcriptases of vpx coding and non-coding lentiviruses. J. Biol. Chem. 2015, 290, 30078–30086. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, G.S.; Booth, H.; Petit, S.J.; Potton, E.; Towers, G.J.; Miller, R.F.; Chain, B.M.; Noursadeghi, M. Adherent human alveolar macrophages exhibit a transient pro-inflammatory profile that confounds responses to innate immune stimulation. PLoS ONE 2012, 7, e40348. [Google Scholar] [CrossRef]
- Descours, B.; Cribier, A.; Chable-Bessia, C.; Ayinde, D.; Rice, G.; Crow, Y.; Yatim, A.; Schwartz, O.; Laguette, N.; Benkirane, M. Samhd1 restricts hiv-1 reverse transcription in quiescent cd4+ t-cells. Retrovirology 2012, 9, 1–8. [Google Scholar] [CrossRef]
- Mlcochova, P.; Winstone, H.; Zuliani-Alvarez, L.; Gupta, R.K. Tlr4-mediated pathway triggers interferon-independent g0 arrest and antiviral samhd1 activity in macrophages. Cell Rep. 2020, 30, 3972–3980.e5. [Google Scholar] [CrossRef]
- Badros, A.; Burger, A.M.; Philip, S.; Niesvizky, R.; Kolla, S.S.; Goloubeva, O.; Harris, C.; Zwiebel, J.; Wright, J.J.; Espinoza-Delgado, I.; et al. Phase i study of vorinostat in combination with bortezomib for relapsed and refractory multiple myeloma. Clin. Cancer. Res. 2009, 15, 5250–5257. [Google Scholar] [CrossRef]
- Dimopoulos, M.; Siegel, D.S.; Lonial, S.; Qi, J.; Hajek, R.; Facon, T.; Rosinol, L.; Williams, C.; Blacklock, H.; Goldschmidt, H.; et al. Vorinostat or placebo in combination with bortezomib in patients with multiple myeloma (vantage 088): A multicentre, randomised, double-blind study. Lancet Oncol. 2013, 14, 1129–1140. [Google Scholar] [CrossRef]
- Halsall, J.A.; Turner, B.M. Histone deacetylase inhibitors for cancer therapy: An evolutionarily ancient resistance response may explain their limited success. Bioessays 2016, 38, 1102–1110. [Google Scholar] [CrossRef]
- West, A.C.; Johnstone, R.W. New and emerging hdac inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef]
- Archin, N.M.; Keedy, K.S.; Espeseth, A.; Dang, H.; Hazuda, D.J.; Margolis, D.M. Expression of latent human immunodeficiency type 1 is induced by novel and selective histone deacetylase inhibitors. AIDS 2009, 23, 1799–1806. [Google Scholar] [CrossRef] [PubMed]
- Wightman, F.; Lu, H.K.; Solomon, A.E.; Saleh, S.; Harman, A.N.; Cunningham, A.L.; Gray, L.; Churchill, M.; Cameron, P.U.; Dear, A.E.; et al. Entinostat is a histone deacetylase inhibitor selective for class 1 histone deacetylases and activates hiv production from latently infected primary t cells. AIDS 2013, 27, 2853–2862. [Google Scholar] [CrossRef] [PubMed]
- Ylisastigui, L.; Archin, N.M.; Lerhrman, G.; Bosch, R.J.; Margolis, D.M. Coaxing hiv-1 from resting cd4 t cells: Histone deacetylase inhibition allows latent viral expression. AIDS 2004, 18, 1101–1108. [Google Scholar] [CrossRef]
- Mlcochova, P.; Caswell, S.J.; Taylor, I.A.; Towers, G.J.; Gupta, R.K. DNA damage induced by topoisomerase inhibitors activates samhd1 and blocks hiv-1 infection of macrophages. EMBO J. 2018, 37, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Jáuregui, P.; Landau, N.R. DNA damage induces a samhd1-mediated block to the infection of macrophages by hiv-1. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef]
- Little, R.F.; Pittaluga, S.; Grant, N.; Steinberg, S.M.; Kavlick, M.F.; Mitsuya, H.; Franchini, G.; Gutierrez, M.; Raffeld, M.; Jaffe, E.S.; et al. Highly effective treatment of acquired immunodeficiency syndrome-related lymphoma with dose-adjusted epoch: Impact of antiretroviral therapy suspension and tumor biology. Blood 2003, 101, 4653–4659. [Google Scholar] [CrossRef]
- Joseph, S.B.; Arrildt, K.T.; Sturdevant, C.B.; Swanstrom, R. Hiv-1 target cells in the cns. J. Neurovirol. 2015, 21, 276–289. [Google Scholar] [CrossRef]
- Wallet, C.; De Rovere, M.; Van Assche, J.; Daouad, F.; De Wit, S.; Gautier, V.; Mallon, P.W.G.; Marcello, A.; Van Lint, C.; Rohr, O.; et al. Microglial cells: The main hiv-1 reservoir in the brain. Front. Cell Infect. Microbiol. 2019, 9, 362. [Google Scholar] [CrossRef]
- Vairo, G.; Royston, A.K.; Hamilton, J.A. Biochemical events accompanying macrophage activation and the inhibition of colony-stimulating factor-1 induced macrophage proliferation by tumor necrosis factor-a, interferon-y, and lipopolysaccharide. J. Cell. Physiol. 1992, 151, 630–641. [Google Scholar] [CrossRef]
- Zhang, K.; Song, F.; Lu, X.; Chen, W.; Huang, C.; Li, L.; Liang, D.; Cao, S.; Dai, H. Microrna-322 inhibits inflammatory cytokine expression and promotes cell proliferation in lps-stimulated murine macrophages by targeting nf-kappab1 (p50). Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef]
- Mytych, J.; Romerowicz-Misielak, M.; Koziorowski, M. Long-term culture with lipopolysaccharide induces dose-dependent cytostatic and cytotoxic effects in thp-1 monocytes. Toxicol. Vitr. 2017, 42, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Thongngarm, T.; Jenkins, J.K.; Ndebele, K.; McMurray, R.W. Estrogen and progesterone modulate monocyte cell cycle progression and apoptosis. Am. J. Reprod. Immunol. 2003, 49, 129–138. [Google Scholar] [CrossRef] [PubMed]
- Vadiveloo, P.K. Macrophages—proliferation, activation, and cell cycle proteins. J. Leukoc. Biol. 1999, 66, 579–582. [Google Scholar] [CrossRef] [PubMed]
- Xaus, J.; Cardó, M.; Valledor, A.F.; Soler, C.; Lloberas, J.; Celada, A. Interferon y induces the expression of p21waf-1 and arrests macrophage cell cycle, preventing induction of apoptosis. Immunity 1999, 11, 103–113. [Google Scholar] [CrossRef]
- Murakami, Y.; Mizoguchi, F.; Saito, T.; Miyasaka, N.; Kohsaka, H. P16(ink4a) exerts an anti-inflammatory effect through accelerated irak1 degradation in macrophages. J. Immunol. 2012, 189, 5066–5072. [Google Scholar] [CrossRef] [PubMed]
- Van Marle, G.; Gill, M.J.; Kolodka, D.; McManus, L.; Grant, T.; Church, D.L. Compartmentalization of the gut viral reservoir in hiv-1 infected patients. Retrovirology 2007, 4, 87. [Google Scholar] [CrossRef] [PubMed]
- Collier, D.A.; Haddow, L.; Brijkumar, J.; Moosa, M.S.; Benjamin, L.; Gupta, R.K. Hiv cerebrospinal fluid escape and neurocognitive pathology in the era of combined antiretroviral therapy: What lies beneath the tip of the iceberg in sub-saharan africa? Brain Sci. 2018, 8, 190. [Google Scholar] [CrossRef]
- Kugathasan, R.; Collier, D.A.; Haddow, L.J.; El Bouzidi, K.; Edwards, S.G.; Cartledge, J.D.; Miller, R.F.; Gupta, R.K. Diffuse white matter signal abnormalities on magnetic resonance imaging are associated with human immunodeficiency virus type 1 viral escape in the central nervous system among patients with neurological symptoms. Clin. Infect. Dis. 2017, 64, 1059–1065. [Google Scholar] [CrossRef]
- Davy, C.; Doorbar, J. G2/m cell cycle arrest in the life cycle of viruses. Virology 2007, 368, 219–226. [Google Scholar] [CrossRef]
- Fan, Y.; Sanyal, S.; Bruzzone, R. Breaking bad: How viruses subvert the cell cycle. Front. Cell. Infect. Microbiol. 2018, 8, 396. [Google Scholar] [CrossRef]
- Nascimento, R.; Costa, H.; Parkhouse, R.M. Virus manipulation of cell cycle. Protoplasma 2012, 249, 519–528. [Google Scholar] [CrossRef] [PubMed]
- Balliet, J.W.; Kolson, D.L.; Eiger, G.; Kim, F.M.; McGann, K.A.; Srinivasan, A.; Collman, R. Distinct effects in primary macrophages and lymphocytes of the human immunodeficiency virus type 1 accessory genes vpr, vpu, and nef: Mutational analysis of a primary hiv-1 isolate. Virology 1994, 200, 623–631. [Google Scholar] [CrossRef] [PubMed]
- Connor, R.I.; Chen, B.K.; Choe, S.; Landau, N.R. Vpr is required for efficient replication of human immunodeficiency virus type-1 in mononuclear phagocytes. Virology 1995, 206, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Fouchier, R.A.M.; Meyer, B.E.; Simon, J.H.M.; Fischer, U.; Albright, A.V.; Gonzáles-Scarano, F.; Malm, M.H. Interaction of the human immunodeficinecy virus type 1 vpr protein with the nuclear pore complex. J. Virol. 1998, 72, 6004–6013. [Google Scholar] [CrossRef]
- Goh, W.C.; Rogel, M.E.; Kinsey, C.M.; Michael, S.F.; Fultz, P.N.; Nowak, M.A.; Hahn, B.H.; Emerman, M. Hiv-1 vpr increases viral expression by manipulation of the cell cycle: A mechanism for selection of vpr in vivo. Nat. Med. 1998, 4, 65–71. [Google Scholar] [CrossRef]
- Groschel, B.; Bushman, F. Cell cycle arrest in g2/m promotes early steps of infection by human immunodeficiency virus. J. Virol. 2005, 79, 5695–5704. [Google Scholar] [CrossRef]
- Gummuluru, S.; Emerman, M. Cell cycle- and vpr-mediated regulation of human immunodeficiency virus type 1 expression in primary and transformed t-cell lines. J. Virol. 1999, 73, 5422–5430. [Google Scholar] [CrossRef]
- Zimmerman, E.S.; Chen, J.; Andersen, J.L.; Ardon, O.; Dehart, J.L.; Blackett, J.; Choudhary, S.K.; Camerini, D.; Nghiem, P.; Planelles, V. Human immunodeficiency virus type 1 vpr-mediated g2 arrest requires rad17 and hus1 and induces nuclear brca1 and gamma-h2ax focus formation. Mol. Cell. Biol. 2004, 24, 9286–9294. [Google Scholar] [CrossRef]
- Laguette, N.; Bregnard, C.; Hue, P.; Basbous, J.; Yatim, A.; Larroque, M.; Kirchhoff, F.; Constantinou, A.; Sobhian, B.; Benkirane, M. Premature activation of the slx4 complex by vpr promotes g2/m arrest and escape from innate immune sensing. Cell 2014, 156, 134–145. [Google Scholar] [CrossRef]
- Chang, J.H.; Kim, J.J.; Choi, J.M.; Lee, J.H.; Cho, Y. Crystal structure of the mus81-eme1 complex. Genes Dev. 2008, 22, 1093–1106. [Google Scholar] [CrossRef]
- Greenwood, E.J.D.; Williamson, J.C.; Sienkiewicz, A.; Naamati, A.; Matheson, N.J.; Lehner, P.J. Promiscuous targeting of cellular proteins by vpr drives systems-level proteomic remodeling in hiv-1 infection. Cell Rep. 2019, 27, 1579–1596.e7. [Google Scholar] [CrossRef] [PubMed]
- Hrecka, K.; Gierszewska, M.; Srivastava, S.; Kozaczkiewicz, L.; Swanson, S.K.; Florens, L.; Washburn, M.P.; Skowronski, J. Lentiviral vpr usurps cul4-ddb1[vprbp] e3 ubiquitin ligase to modulate cell cycle. Proc. Natl. Acad. Sci. USA 2007, 104, 11778–11783. [Google Scholar] [CrossRef] [PubMed]
- Le Rouzic, E.; Belaidouni, N.; Estrabaud, E.; Morel, M.; Rain, J.C.; Transy, C.; Margottin-Goguet, F. Hiv1 vpr arrests the cell cycle by recruiting dcaf1/vprbp, a receptor of the cul4-ddb1 ubiquitin ligase. Cell Cycle 2007, 6, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Ehrlich, E.; Yu, X.F. Ddb1 and cul4a are required for human immunodeficiency virus type 1 vpr-induced g2 arrest. J. Virol. 2007, 81, 10822–10830. [Google Scholar] [CrossRef]
- Wen, X.; Duus, K.M.; Friedrich, T.D.; de Noronha, C.M. The hiv1 protein vpr acts to promote g2 cell cycle arrest by engaging a ddb1 and cullin4a-containing ubiquitin ligase complex using vprbp/dcaf1 as an adaptor. J. Biol. Chem. 2007, 282, 27046–27057. [Google Scholar] [CrossRef]
- Yan, J.; Shun, M.C.; Zhang, Y.; Hao, C.; Skowronski, J. Hiv-1 vpr counteracts hltf-mediated restriction of hiv-1 infection in t cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9568–9577. [Google Scholar] [CrossRef]
- Izumi, T.; Io, K.; Matsui, M.; Shirakawa, K.; Shinohara, M.; Nagai, Y.; Kawahara, M.; Kobayashi, M.; Kondoh, H.; Misawa, N.; et al. Hiv-1 viral infectivity factor interacts with tp53 to induce g2 cell cycle arrest and positively regulate viral replication. Proc. Natl. Acad. Sci. USA 2010, 107, 20798–20803. [Google Scholar] [CrossRef]
- Stopak, K.; de Noronha, C.M.; Yonemoto, W.; Greene, W.C. Hiv-1 vif blocks the antiviral activity of apobec3g by impairing both its translation and intracellular stability. Mol. Cell 2003, 12, 591–601. [Google Scholar] [CrossRef]
- Wang, J.; Schackelford, J.M.; Casella, C.R.; Shivers, D.K.; Rapport, E.L.; Liu, B.; Yu, X.F.; Finkel, T.H. The vif accessory protein alters the cell cycle of human immunodeficiency virus type 1 infected cells. Virology 2007, 359, 243–252. [Google Scholar] [CrossRef]
- DeHart, J.L.; Bosque, A.; Harris, R.S.; Planelles, V. Human immunodeficiency virus type 1 vif induces cell cycle delay via recruitment of the same e3 ubiquitin ligase complex that targets apobec3 proteins for degradation. J. Virol. 2008, 82, 9265–9272. [Google Scholar] [CrossRef]
- Sakai, K.; Dimas, J.; Lenardo, M.J. The vif and vpr accessory proteins independently cause hiv-1-induced t cell cytopathicity and cell cycle arrest. Proc. Natl. Acad. Sci. USA 2006, 103, 3369–3374. [Google Scholar] [CrossRef] [PubMed]
- Zhao, K.; Du, J.; Rui, Y.; Zheng, W.; Kang, J.; Hou, J.; Wang, K.; Zhang, W.; Simon, V.A.; Yu, X.F. Evolutionarily conserved pressure for the existence of distinct g2/m cell cycle arrest and a3h inactivation functions in hiv-1 vif. Cell Cycle 2015, 14, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Salamango, D.J.; Ikeda, T.; Moghadasi, S.A.; Wang, J.; McCann, J.L.; Serebrenik, A.A.; Ebrahimi, D.; Jarvis, M.C.; Brown, W.L.; Harris, R.S. Hiv-1 vif triggers cell cycle arrest by degrading cellular ppp2r5 phospho-regulators. Cell Rep. 2019, 29, 1057–1065.e4. [Google Scholar] [CrossRef]
- Greenwood, E.J.; Matheson, N.J.; Wals, K.; van den Boomen, D.J.; Antrobus, R.; Williamson, J.C.; Lehner, P.J. Temporal proteomic analysis of hiv infection reveals remodelling of the host phosphoproteome by lentiviral vif variants. Elife 2016, 5, e18296. [Google Scholar] [CrossRef] [PubMed]
- Foley, E.A.; Maldonado, M.; Kapoor, T.M. Formation of stable attachments between kinetochores and microtubules depends on the b56-pp2a phosphatase. Nat. Cell Biol. 2011, 13, 1265–1271. [Google Scholar] [CrossRef]
- Grallert, A.; Boke, E.; Hagting, A.; Hodgson, B.; Connolly, Y.; Griffiths, J.R.; Smith, D.L.; Pines, J.; Hagan, I.M. A pp1-pp2a phosphatase relay controls mitotic progression. Nature 2015, 517, 94–98. [Google Scholar] [CrossRef]
- Nasa, I.; Kettenbach, A.N. Coordination of protein kinase and phosphoprotein phosphatase activities in mitosis. Front. Cell Dev. Biol. 2018, 6, 30. [Google Scholar] [CrossRef]
- Vallardi, G.; Allan, L.A.; Crozier, L.; Saurin, A.T. Division of labour between pp2a-b56 isoforms at the centromere and kinetochore. Elife 2019, 8, 8. [Google Scholar] [CrossRef]
- Marelli, S.; Williamson, J.C.; Protasio, A.V.; Naamati, A.; Greenwood, E.J.D.; Deane, J.E.; Lehner, P.J.; Matheson, N.J. Antagonism of pp2a is an independent and conserved function of hiv-1 vif and causes cell cycle arrest. bioRxiv 2019, 9, e53036. [Google Scholar] [CrossRef]
- Naamati, A.; Williamson, J.C.; Greenwood, E.J.; Marelli, S.; Lehner, P.J.; Matheson, N.J. Functional proteomic atlas of hiv infection in primary human cd4+ t cells. Elife 2019, 8, 8. [Google Scholar] [CrossRef]
- Sakai, K.; Barnitz, R.A.; Chaigne-Delalande, B.; Bidere, N.; Lenardo, M.J. Human immunodeficiency virus type 1 vif causes dysfunction of cdk1 and cyclinb1: Implications for cell cycle arrest. Virol. J. 2011, 8, 219. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Reuschel, E.L.; Shackelford, J.M.; Jeang, L.; Shivers, D.K.; Diehl, J.A.; Yu, X.F.; Finkel, T.H. Hiv-1 vif promotes the g(1)- to s-phase cell-cycle transition. Blood 2011, 117, 1260–1269. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Shun, M.C.; Hao, C.; Zhang, Y.; Qian, J.; Hrecka, K.; DeLucia, M.; Monnie, C.; Ahn, J.; Skowronski, J. Hiv-1 vpr reprograms clr4dcaf1 e3 ubiquitinn ligase to antagonize exonuclease 1-mediated restriction of hiv-1 infection. mBio 2018, 9, 1–15. [Google Scholar] [CrossRef] [PubMed]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferreira, I.A.T.M.; Porterfield, J.Z.; Gupta, R.K.; Mlcochova, P. Cell Cycle Regulation in Macrophages and Susceptibility to HIV-1. Viruses 2020, 12, 839. https://doi.org/10.3390/v12080839
Ferreira IATM, Porterfield JZ, Gupta RK, Mlcochova P. Cell Cycle Regulation in Macrophages and Susceptibility to HIV-1. Viruses. 2020; 12(8):839. https://doi.org/10.3390/v12080839
Chicago/Turabian StyleFerreira, Isabella A. T. M., J. Zachary Porterfield, Ravindra K. Gupta, and Petra Mlcochova. 2020. "Cell Cycle Regulation in Macrophages and Susceptibility to HIV-1" Viruses 12, no. 8: 839. https://doi.org/10.3390/v12080839
APA StyleFerreira, I. A. T. M., Porterfield, J. Z., Gupta, R. K., & Mlcochova, P. (2020). Cell Cycle Regulation in Macrophages and Susceptibility to HIV-1. Viruses, 12(8), 839. https://doi.org/10.3390/v12080839