BKTyper: Free Online Tool for Polyoma BK Virus VP1 and NCCR Typing

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
3. Results
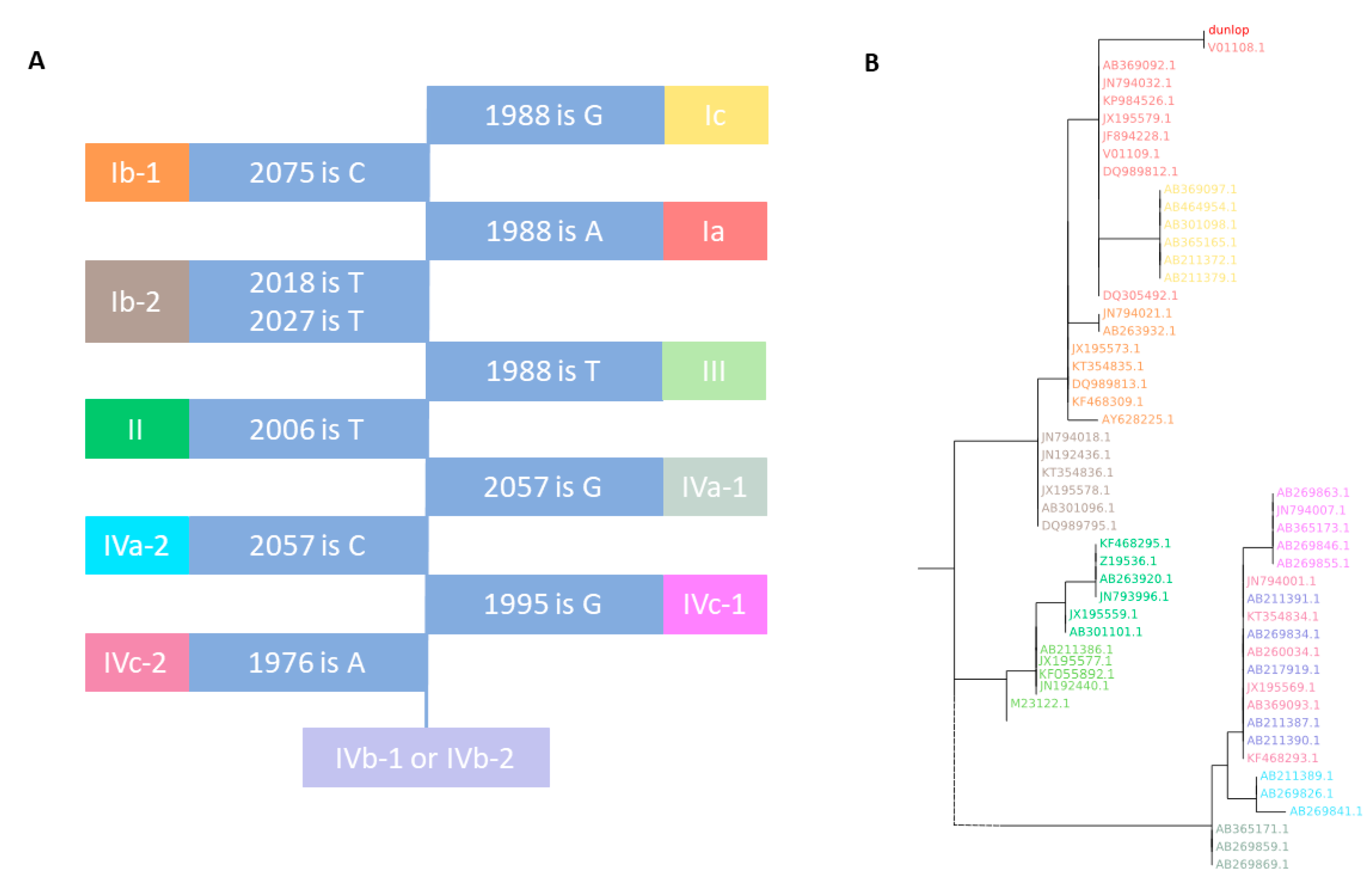
3.1. VP1 Genotyping
3.1.1. VP1 BKTGR (BK Typing and Grouping Region)
3.1.2. BKTGR Phylogeny
3.1.3. VP1 Genotyping Validation
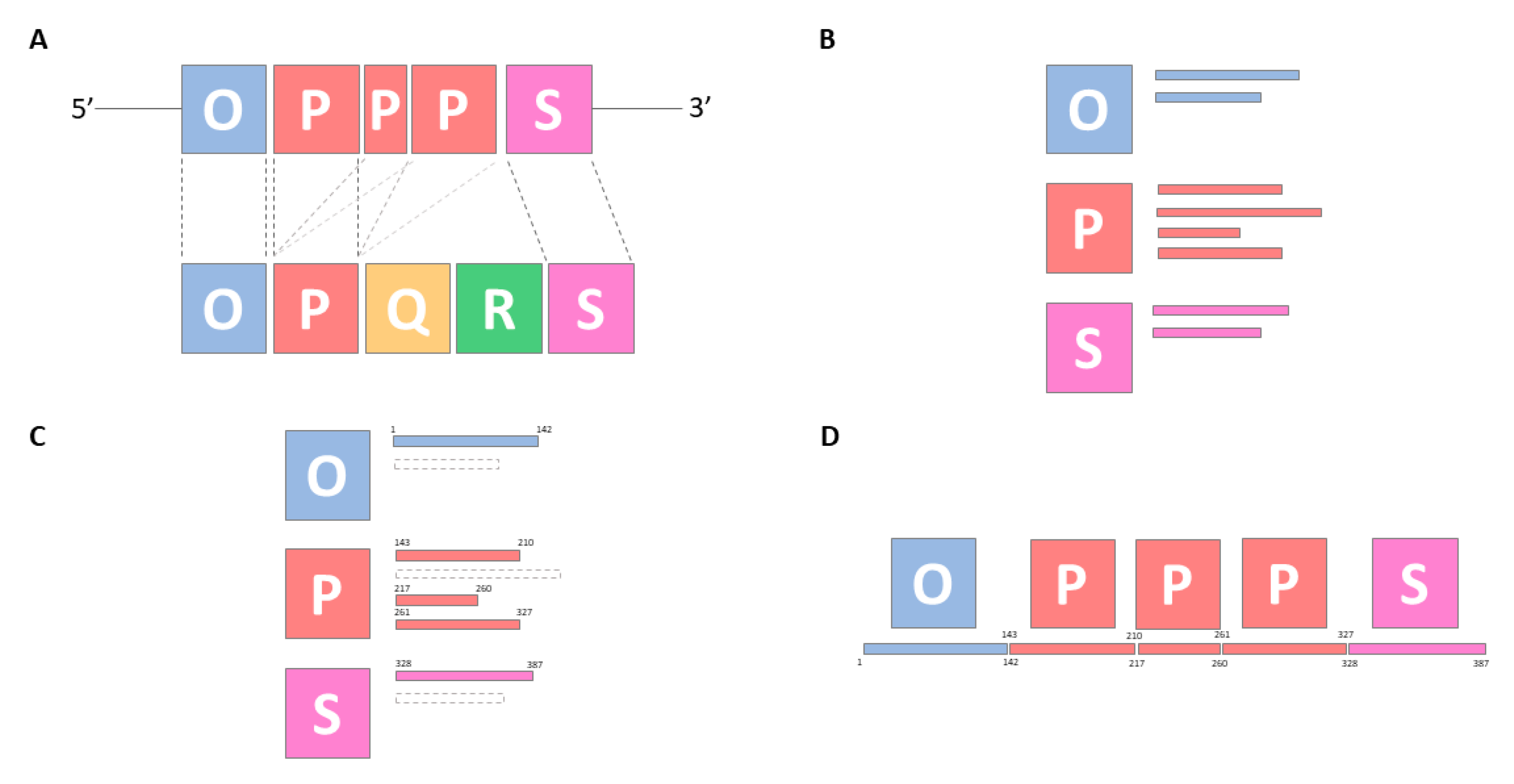
3.2. NCCR Typing
3.2.1. Defining a Canon for the Archetypical OPQRS Blocks
3.2.2. NCCR Typing Based on Local Alignment
3.2.3. NCCR Typing Validation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A
| Open nucleotide sequence in fasta format: |
| Locally align sequence against VP1 gene from BKPyV virus Dunlop strain using Needleman–Wunch algorithm: |
| Gap open penalty of 10 |
| Gap extension penalty of 0.5 |
| Trim alignment: |
| From beginning until first position without gap |
| From end until first position without gap |
| Reallocate Dunlop coordinates {1976, 1988, 1995, 2006, 2018, 2057, 2075} to alignment coordinates: |
| For each Dunlop coordinate: |
| Define specific motif in Dunlop sequence |
| Store Dunlop coordinate and specific motif |
| Search specific motifs by coordinate order in alignment: |
| If match: |
| Compare the query position of the first position in Dunlop motif match: |
| If nucleotide in subject (Dunlop-motif-1988) is G: |
| Then subgroup is Ic |
| If nucleotide in subject (Dunlop-motif-2075) is C: |
| Then subgroup is Ib-1 |
| If nucleotide in subject (Dunlop-motif-1988) is A: |
| Then subgroup is Ia |
| If nucleotide in subject (Dunlop-motif-2018) is T AND |
| nucleotide in subject (Dunlop-motif-2027) is T: |
| Then subgroup is Ib-2 |
| If nucleotide in subject (Dunlop-motif-1988) is T: |
| Then subgroup is III |
| If nucleotide in subject (Dunlop-motif-2006) is T: |
| Then subgroup is II |
| If nucleotide in subject (Dunlop-motif-2057) is G: |
| Then subgroup is IVa-1 |
| If nucleotide in subject (Dunlop-motif-2057) is C: |
| Then subgroup is IVa-2 |
| If nucleotide in subject (Dunlop-motif-1995) is G: |
| Then subgroup is IVc-1 |
| If nucleotide in subject (Dunlop-motif-1976) is A: |
| Then subgroup is IVc-2 |
| If not any of the above: |
| Then subgroup is IVb-1 or IVb-2 |
| Report BKPyV subgroup and nucleotide in subject (Dunlop-motif-1976, Dunlop-motif-1988, Dunlop-motif-1995, Dunlop-motif-2006, Dunlop-motif-2018, Dunlop-motif-2057, Dunlop-motif-2075). |
Appendix B
| Open nucleotide sequence in fasta format: |
| Locally align sequence against NCCR BK virus archetypes using blastn: |
| Word size of 12 |
| Minimum e-value of 0.05 |
| Minimum percentage of identity of 75% |
| Read alignment: |
| Order ascendingly query start sequence position: |
| By block type: |
| By query start and end sequence positions: |
| Keep the longest alignment |
| Report the start and end block positions |
| Report BK NCCR organization and coordinates of each block |
References
- Gardner, S.; Field, A.; Coleman, D.; Hulme, B. New human papovavirus (B.K.) isolated from urine after renal transplantation. Lancet 1971, 297, 1253–1257. [Google Scholar] [CrossRef]
- Krumbholz, A.; Bininda-Emonds, O.R.P.; Wutzler, P.; Zell, R. Evolution of four BK virus subtypes. Infect. Genet. Evol. 2008, 8, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.H.; Knowles, W.; Dickenmann, M.; Passweg, J.; Klimkait, T.; Mihatsch, M.J.; Steiger, J. Prospective Study of Polyomavirus Type BK Replication and Nephropathy in Renal-Transplant Recipients. N. Engl. J. Med. 2002, 347, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Bethge, T.; Hachemi, H.A.; Manzetti, J.; Gosert, R.; Schaffner, W.; Hirsch, H.H. Sp1 Sites in the Noncoding Control Region of BK Polyomavirus Are Key Regulators of Bidirectional Viral Early and Late Gene Expression. J. Virol. 2015, 89, 3396–3411. [Google Scholar] [CrossRef]
- Carr, M.J.; McCormack, G.P.; Mutton, K.J.; Crowley, B. Unique BK virus non-coding control region (NCCR) variants in hematopoietic stem cell transplant recipients with and without hemorrhagic cystitis. J. Med. Virol. 2006, 78, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Barcena-Panero, A.; Echevarria, J.E.; Van Ghelue, M.; Fedele, G.; Royuela, E.; Gerits, N.; Moens, U. BK polyomavirus with archetypal and rearranged non-coding control regions is present in cerebrospinal fluids from patients with neurological complications. J. Gen. Virol. 2012, 93, 1780–1794. [Google Scholar] [CrossRef] [PubMed]
- Burger-Calderon, R.; Ramsey, K.J.; Dolittle-Hall, J.M.; Seaman, W.T.; Jeffers-Francis, L.K.; Tesfu, D.; Nickeleit, V.; Webster-Cyriaque, J. Distinct BK polyomavirus non-coding control region (NCCR) variants in oral fluids of HIV-associated Salivary Gland Disease patients. Virology 2016, 493, 255–266. [Google Scholar] [CrossRef]
- Gosert, R.; Rinaldo, C.H.; Funk, G.A.; Egli, A.; Ramos, E.; Drachenberg, C.B.; Hirsch, H.H. Polyomavirus BK with rearranged noncoding control region emerge in vivo in renal transplant patients and increase viral replication and cytopathology. J. Exp. Med. 2008, 205, 841–852. [Google Scholar] [CrossRef]
- Jin, L. Molecular Methods for Identification and Genotyping of BK Virus. In SV40 Protocols; Humana Press: New Jersey, NJ, USA, 2015; Volume 165, pp. 33–48. [Google Scholar]
- Ranjan, R.; Rani, A.; Brennan, D.C.; Finn, P.W.; Perkins, D.L. Complete Genome Sequence of BK Polyomavirus Subtype Ib-1 Detected in a Kidney Transplant Patient with BK Viremia Using Shotgun Sequencing. Genome Announc. 2017, 5. [Google Scholar] [CrossRef]
- Zhong, S.; Randhawa, P.S.; Ikegaya, H.; Chen, Q.; Zheng, H.-Y.; Suzuki, M.; Takeuchi, T.; Shibuya, A.; Kitamura, T.; Yogo, Y. Distribution patterns of BK polyomavirus (BKV) subtypes and subgroups in American, European and Asian populations suggest co-migration of BKV and the human race. J. Gen. Virol. 2009, 90, 144–152. [Google Scholar] [CrossRef]
- Nishimoto, Y.; Zheng, H.-Y.; Zhong, S.; Ikegaya, H.; Chen, Q.; Sugimoto, C.; Kitamura, T.; Yogo, Y. An Asian Origin for Subtype IV BK Virus Based on Phylogenetic Analysis. J. Mol. Evol. 2007, 65, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Morel, V.; Martin, E.; François, C.; Helle, F.; Faucher, J.; Mourez, T.; Choukroun, G.; Duverlie, G.; Castelain, S.; Brochot, E. A Simple and Reliable Strategy for BK Virus Subtyping and Subgrouping. J. Clin. Microbiol. 2017, 55, 1177–1185. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, S.; Chakraborty, T. High Reactivation of BK Virus Variants in Asian Indians with Renal Disorders and During Pregnancy. Virus Genes 2004, 28, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Egli, A.; Infanti, L.; Dumoulin, A.; Buser, A.; Samaridis, J.; Stebler, C.; Gosert, R.; Hirsch, H.H. Prevalence of Polyomavirus BK and JC Infection and Replication in 400 Healthy Blood Donors. J. Infect. Dis. 2009, 199, 837–846. [Google Scholar] [CrossRef]
- Helle, F.; Brochot, E.; Handala, L.; Martin, E.; Castelain, S.; Francois, C.; Duverlie, G. Biology of the BKPyV: An Update. Viruses 2017, 9, 327. [Google Scholar] [CrossRef]
- Wang, R.Y.L.; Li, Y.-J.; Lee, W.-C.; Wu, H.-H.; Lin, C.-Y.; Lee, C.-C.; Chen, Y.-C.; Hung, C.-C.; Yang, C.-W.; Tian, Y.-C. The association between polyomavirus BK strains and BKV viruria in liver transplant recipients. Sci. Rep. 2016, 6, 28491. [Google Scholar] [CrossRef]
- Sharma, P.M.; Gupta, G.; Vats, A.; Shapiro, R.; Randhawa, P.S. Polyomavirus BK non-coding control region rearrangements in health and disease. J. Med. Virol. 2007, 79, 1199–1207. [Google Scholar] [CrossRef]
- Anzivino, E.; Bellizzi, A.; Mitterhofer, A.; Tinti, F.; Barile, M.; Colosimo, M.; Fioriti, D.; Mischitelli, M.; Chiarini, F.; Ferretti, G.; et al. Early monitoring of the human polyomavirus BK replication and sequencing analysis in a cohort of adult kidney transplant patients treated with basiliximab. Virol. J. 2011, 8, 407. [Google Scholar] [CrossRef]
- Liimatainen, H.; Weseslindtner, L.; Strassl, R.; Aberle, S.W.; Bond, G.; Auvinen, E. Next-generation sequencing shows marked rearrangements of BK polyomavirus that favor but are not required for polyomavirus-associated nephropathy. J. Clin. Virol. 2020, 122, 104215. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Pritchard, L.; White, J.A.; Birch, P.R.J.; Toth, I.K. GenomeDiagram: A python package for the visualization of large-scale genomic data. Bioinformatics 2006, 22, 616–617. [Google Scholar] [CrossRef]
- van der Walt, S.; Colbert, S.C.; Varoquaux, G. The NumPy Array: A Structure for Efficient Numerical Computation. Comput. Sci. Eng. 2011, 13, 22–30. [Google Scholar] [CrossRef]
- McKinney, W. Data Structures for Statistical Computing in Python. In Proceedings of the 9th Python in Science Conference, Austin, TX, USA, 28 June–3 July 2010; 445, pp. 56–61. [Google Scholar]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST plus: Architecture and applications. BMC Bioinform. 2009, 10, 1. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Schmidt, H.A.; Chernomor, O.; Schrempf, D.; Woodhams, M.D.; von Haeseler, A.; Lanfear, R. IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era. Mol. Biol. Evol. 2020, 37, 1530–1534. [Google Scholar] [CrossRef]
- Needleman, S.B.; Wunsch, C.D. A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 1970, 48, 443–453. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Markowitz, R.B.; Dynan, W.S. Binding of cellular proteins to the regulatory region of BK virus DNA. J. Virol. 1988, 62, 3388–3398. [Google Scholar] [CrossRef]
- Moens, U.; Van Ghelue, M. Polymorphism in the genome of non-passaged human polyomavirus BK: Implications for cell tropism and the pathological role of the virus. Virology 2005, 331, 209–231. [Google Scholar] [CrossRef] [PubMed]
- Moens, U.; Johansen, T.; Johnsen, J.I.; Seternes, O.M.; Traavik, T. Noncoding control region of naturally occurring BK virus variants: Sequence comparison and functional analysis. Virus Genes 1995, 10, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Burger-Calderon, R.; Madden, V.; Hallett, R.A.; Gingerich, A.D.; Nickeleit, V.; Webster-Cyriaque, J. Replication of Oral BK Virus in Human Salivary Gland Cells. J. Virol. 2014, 88, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Yang, R.; Wu, R. BK virus DNA: Complete nucleotide sequence of a human tumor virus. Science 1979, 206, 456–462. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCCR Block | Sequence |
|---|---|
| O | TTTTGCAAAAATTGCAAAAGAATAGGGATTTCCCCAAATAGTTTTGCTAGGCCTCAGAAAAAGCCTCCACACCCTTACTACTTGAGAGAAAGGGTGGAGGCAGAGGCGGCCTCGGCCTCTTATATATTATAAAAAAAAAGGC |
| P | CACAGGGAGGAGCTGCTTACCCATGGAATGCAGCCAAACCATGACCTCAGGAAGGAAAGTGCATGACT |
| Q | GGGCAGCCAGCCAGTGGCAGTTAATAGTGAAACCCCGCC |
| R | CCTGAAATTCTCAAATAAACACAAGAGGAAGTGGAAACTGGCCAAAGGAGTGGAAAGCAGCCA |
| S | GACAGACATGTTTTGCGAGCCTAGGAATCTTGGCCTTGTCCCCAGTTAAACTGGACAAAGGCC |
| Genome Coordinates | VP1 Coordinates | Conserved Motif |
|---|---|---|
| 1989 | 426 | AAAACCTAT |
| 2076 | 513 | AAGTAC |
| 2019 | 456 | CTTTGCTG |
| 2028 | 465 | AGGTGGAGAA |
| 2007 | 444 | TAATTTCCACTTCTTTG |
| 2058 | 495 | GCTAATGAATTACAG |
| 1996 | 433 | ATTCAAGGCAGTAATTT |
| 1977 | 414 | GCATGGTGGAGGAAA |
| Transcriptional Binding Sites | Motif |
|---|---|
| Promoter IL-6 gene | TTCC |
| T-Antigen | GCCTC or GCCCC |
| NF-1 | TCCA or TGGCCTTGTCCCCAG |
| Polyomavirus enhancer B | AGAGG |
| SP-1 | AGGCGG |
| Unknown JC polyomavirus binding factor (JVC) | GGGAGGAG |
| Cytomegalovirus immediate early promoter (CMV IE-1) | GGAAAG |
| NFkB | GTGAAACCCC |
| SV40 enhancer-core | TGGAAAG |
| CRE | TGACCTCA |
| GRE | TGTCCC |
| Murine Thy-1 | AGGC |
| TATA box | TATAA |
| Transcription factor Late SV40 (LSF) | CCCGCC |
| Percentage | N | NCCR | VP1 |
|---|---|---|---|
| 24.53 | 78 | OPQRS | Ic |
| 16.04 | 51 | OPQRS | Ib-2 |
| 12.26 | 39 | OPQRS | Ib-1 |
| 9.12 | 29 | OPQRS | IVc-2 |
| 5.35 | 17 | OPQRS | IVc-1 |
| 5.35 | 17 | OPQRS | IVb-1,2 |
| 4.09 | 13 | NA | Ib-2 |
| 3.14 | 10 | OPQRS | Ia |
| 2.52 | 8 | OPQRS | III |
| 2.52 | 8 | OPPQRS | Ic |
| 2.20 | 7 | OPQRS | IVa-2 |
| 1.57 | 5 | OPQRS | IVa-1 |
| 1.57 | 5 | OPQRS | II |
| 1.26 | 4 | OPQROPQRS | Ic |
| 1.26 | 4 | OPQPQS | Ia |
| 1.26 | 4 | OPOPQRS | Ic |
| 1.26 | 4 | NA | Ia |
| 0.94 | 3 | NA | Ib-1 |
| 0.63 | 2 | OPPQRS | Ib-1 |
| 0.63 | 2 | OPPPS | Ia |
| 0.31 | 1 | OPS | Ia |
| 0.31 | 1 | OPQSPQS | Ia |
| 0.31 | 1 | OPQRSRS | IVb-1.2 |
| 0.31 | 1 | OPQQRS | Ib-1 |
| 0.31 | 1 | OPQPQPQS | Ia |
| 0.31 | 1 | OPQOPQRS | Ic |
| 0.31 | 1 | OPPQR | IVb-1.2 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martí-Carreras, J.; Mineeva-Sangwo, O.; Topalis, D.; Snoeck, R.; Andrei, G.; Maes, P. BKTyper: Free Online Tool for Polyoma BK Virus VP1 and NCCR Typing. Viruses 2020, 12, 837. https://doi.org/10.3390/v12080837
Martí-Carreras J, Mineeva-Sangwo O, Topalis D, Snoeck R, Andrei G, Maes P. BKTyper: Free Online Tool for Polyoma BK Virus VP1 and NCCR Typing. Viruses. 2020; 12(8):837. https://doi.org/10.3390/v12080837
Chicago/Turabian StyleMartí-Carreras, Joan, Olga Mineeva-Sangwo, Dimitrios Topalis, Robert Snoeck, Graciela Andrei, and Piet Maes. 2020. "BKTyper: Free Online Tool for Polyoma BK Virus VP1 and NCCR Typing" Viruses 12, no. 8: 837. https://doi.org/10.3390/v12080837
APA StyleMartí-Carreras, J., Mineeva-Sangwo, O., Topalis, D., Snoeck, R., Andrei, G., & Maes, P. (2020). BKTyper: Free Online Tool for Polyoma BK Virus VP1 and NCCR Typing. Viruses, 12(8), 837. https://doi.org/10.3390/v12080837