Bioinformatics Pipeline for Human Papillomavirus Short Read Genomic Sequences Classification Using Support Vector Machine

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Reference Genome Sequences
2.2. Epidemiological and Control Samples
2.3. Sequencing
2.4. The Typing Algorithm: Outline and Initial Steps
- total number of aligned read pairs mapped to a given HPV genome
- average depth of alignment of the read pairs to the given HPV genome
- average coverage of the given HPV genome by aligned read pairs
- rate of distinct read pairs
2.5. Pattern Recognition Module of the HPV Typing Pipeline
2.6. Validation of Pipeline Classification
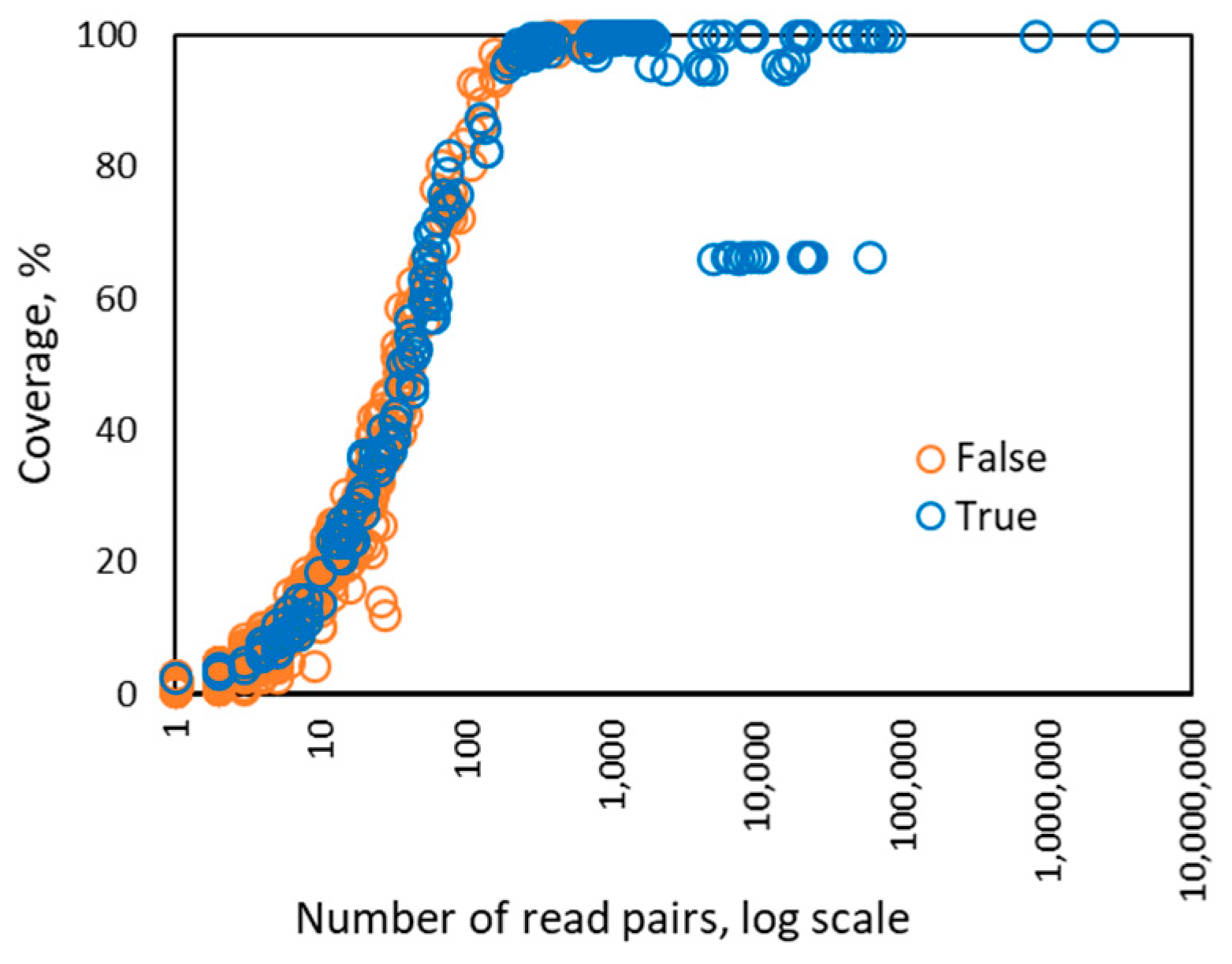
3. Results
3.1. Automated HPV Typing by a Bioinformatics Pipeline: Control/Designed Samples (n = 122)
3.2. Automated HPV Typing by Bioinformatics Pipeline: Epidemiological Samples (n = 261)
4. Discussion
- i.
- HPV read pairs from one sample incorrectly assigned to another sample (index swapping);
- ii.
- read pairs similar to some HPV genomes but originated from unknown sample contamination.
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Li, T.; Unger, E.R.; Batra, D.; Sheth, M.; Steinau, M.; Jasinski, J.; Jones, J.; Rajeevan, M.S. Universal Human Papillomavirus Typing Assay: Whole-Genome Sequencing following Target Enrichment. J. Clin. Microbiol. 2017, 55, 811–823. [Google Scholar] [CrossRef]
- Li, T.; Unger, E.R.; Rajeevan, M.S. Universal human papillomavirus typing by whole genome sequencing following target enrichment: Evaluation of assay reproducibility and limit of detection. BMC Genom. 2019, 20, 231. [Google Scholar] [CrossRef] [PubMed]
- Van Doorslaer, K.; Tan, Q.; Xirasagar, S.; Bandaru, S.; Gopalan, V.; Mohamoud, Y.; Huyen, Y.; McBride, A.A. The Papillomavirus Episteme: A central resource for papillomavirus sequence data and analysis. Nucleic. Acids Res. 2013, 41, D571–D578. [Google Scholar] [CrossRef] [PubMed]
- The NCBI Handbook (Internet). Bethesda (MD): National Library of Medicine (US): National Center for Biotechnology Information. 2019. Available online: http://www.ncbi.nlm.nih.gov/books/NBK21101 (accessed on 3 June 2020).
- Schubert, M.; Lindgreen, S.; Orlando, L. AdapterRemoval v2: Rapid adapter trimming, identification, and read merging. BMC Res. Notes 2016, 9, 88. [Google Scholar] [CrossRef] [PubMed]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.-C.; Lin, C.-J. LIBSVM: A library for support vector machines. ACM Trans. Intell. Syst. Technol. 2011, 2, 27. [Google Scholar] [CrossRef]
- Kircher, M.; Sawyer, S.; Meyer, M. Double indexing overcomes inaccuracies in multiplex sequencing on the Illumina platform. Nucleic. Acids Res. 2012, 40, e3. [Google Scholar] [CrossRef] [PubMed]
- QuickCalcs. Available online: https://www.graphpad.com/quickcalcs/ (accessed on 3 June 2020).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set 1 (n = 32) | Data Set 2 (n = 64) | ||||
|---|---|---|---|---|---|
| Sample ID | Sample Description | Input/Reaction | Experiment 1 Sample ID | Sample Description | Input/Reaction |
| 1 | HPV-plasmid-45 | 50,000 copies | 1 | Pool of HPV plasmid-11,16,31,45,52 | 625 copies |
| 2 | HPV-plasmid-58 | 50,000 copies | 2 | Pool of HPV plasmid-11,16,31,45,52 | 125 copies |
| 3 | HPV-plasmid-31 | 50,000 copies | 3 | Pool of HPV plasmid-11,16,31,45,52 | 25 copies |
| 4 | HPV-plasmid-33 | 50,000 copies | 4 | Pool of HPV plasmid-11,16,31,45,52 | 5 copies |
| 5 | HPV-plasmid-52 | 50,000 copies | 5 | Pool of HPV plasmid-11,16,31,45,52 | 1 copy |
| 6 | HPV-plasmid-6 | 50,000 copies | 6 | Pool of HPV plasmid-6,18,33,58 | 625 copies |
| 7 | HPV-plasmid-18 | 50,000 copies | 7 | Pool of HPV plasmid-6,18,33,58 | 125 copies |
| 8 | HPV-plasmid-11 | 50,000 copies | 8 | Pool of HPV plasmid-6,18,33,58 | 25 copies |
| 9 | H2O (HPV negative) | 0 ng | 9 | Pool of HPV plasmid-6,18,33,58 | 5 copies |
| 10 | Placenta (HPV negative) | 100 ng | 10 | Pool of HPV plasmid-6,18,33,58 | 1 copy |
| 11 | CaSki (HPV-16) | 100 ng | 11 | HPV plasmid-16 | 10,000 copies |
| 12 | CaSki (HPV-16) | 10 ng | 12 | HPV plasmid-18 | 10,000 copies |
| 13 | SiHa (HPV-16) | 100 ng | 13 | H2O (HPV negative) | 0 ng |
| 14 | SiHa (HPV-16) | 10 ng | 14 | Placenta (HPV negative) | 100 ng |
| 15 | HeLa (HPV-18) | 100 ng | 15 | SiHa (HPV-16) | 10 ng |
| 16 | HeLa (HPV-18) | 10 ng | 16 | HeLa (HPV-18) | 10 ng |
| 17–31 | Epidemiological samples (50 extracts from male genital swab) | 100 ng | 17–32 | Replicate of 1–16 | |
| 32 | HPV-plasmid-16 | 50,000 copies | Experiment 2 Sample ID | Repeated as in Experiment 1 | |
| Data Set 3 (n = 64) | Data Set 4 (n = 224) | ||||
| Sample ID | Sample Description | Input/Reaction | Sample ID | Sample Description | Input/Reaction |
| 15 | H2O (HPV negative) | 0 ng | 15 | H2O (HPV negative) | 0 ng |
| 16 | SiHa (HPV-16) | 10 ng | 16 | SiHa (HPV-16) | 10 ng |
| 31 | Placenta (HPV negative) | 100 ng | 31 | Placenta (HPV negative) | 100 ng |
| 32 | Pool of HPV plasmid-11,16,31,45,52 | 625 copies | 32 | HeLa (HPV-18) | 10 ng |
| 44 | Pool of HPV plasmid-5,8,23,36 | 625 copies | 47 | H2O (HPV negative) | 0 ng |
| 45 | Pool of HPV plasmid-6,16,20,24,36,58 | 625 copies | 48 | SiHa (HPV-16) | 10 ng |
| 46 | Pool of HPV plasmid-5,11,15,45,52 | 625 copies | 63 | Placenta (HPV negative) | 100 ng |
| 47 | H2O (HPV negative) | 0 ng | 64 | HeLa (HPV-18) | 10 ng |
| 48 | SiHa (HPV-16) | 10 ng | IDs: 1–14, 17–30, 33–46, 49–62 | Epidemiological samples (56 extracts from cervical cells in PreservCyt) | Total 196 epidemiological samples in data set 4; sample input ranged from 25–100 ng |
| 61 | Pool of HPV plasmid-15,20,24,48 | 625 copies | IDs 65–128 * | The same order as in 1–64 | |
| 62 | Pool of HPV plasmid-8,18,23,31,33,48,53 | 625 copies | IDs 129–192 * | The same order as in 1–64 | |
| 63 | Placenta (HPV negative) | 100 ng | IDs 193–224 * | The same order as in 1–32 | |
| 64 | Pool of HPV plasmid-6,18,33,53,58 | 625 copies | |||
| IDs: 1–14, 17–30, 33–43, 49–60 | Epidemiological samples (50 extracts from male genital swab) | 10–100 ng | |||
| HPV Types in Samples | HPV Copy Number in Samples | ||||
|---|---|---|---|---|---|
| 625 | 125 | 25 | 5 | 1 | |
| HPV-11 | 4 | 4 | 4 | 2 | 0 |
| HPV-16 | 4 | 4 | 4 | 3 | 0 |
| HPV-31 | 4 | 4 | 4 | 2 | 0 |
| HPV-45 | 4 | 4 | 4 | 4 | 0 |
| HPV-52 | 4 | 4 | 4 | 2 | 0 |
| False positives | − | − | − | − | − |
| HPV-6 | 4 | 4 | 4 | 0 | 0 |
| HPV-18 | 4 | 4 | 4 | 1 | 0 |
| HPV-33 | 4 | 4 | 4 | 3 | 0 |
| HPV-58 | 4 | 4 | 4 | 3 | 0 |
| False positives | − | − | − | − | − |
| Dataset ID | Ture HPV Instances | Correctly Detected | False Positives |
|---|---|---|---|
| Set 1 | 15 | 15 | 0 |
| Set 2 | 196 | 144 | 0 |
| Set 3 | 38 | 38 | 2 |
| Set 4 | 14 | 14 | 9 |
| Total | 263 | 211 | 11 |
| Data Set | LA Results | Total | Agreement (%, K) | Sensitivity (%) | Specificity (%) | |||
|---|---|---|---|---|---|---|---|---|
| + | − | |||||||
| Set 1 (n = 15) | NGS Results | + | 48 | 11 | 59 | 93.33 (518/555); k = 0.684 (95% CI 0.590–0.779) (substantial) p = 0.0214 | 65 (48/74) | 97.7 (470/481) |
| − | 26 | 470 | 496 | |||||
| Total | 74 | 481 | 555 | |||||
| Set 3 (n = 50) | NGS Results | + | 43 | 21 | 64 | 97.5 (7072/7252); k = 0.803 (95% CI 0.775–0.831) (substantial) p = 0.1172 | 78 (43/55) | 98.8 (1774/1795) |
| − | 12 | 1774 | 1786 | |||||
| Total | 55 | 1795 | 1850 | |||||
| Set 4 (n = 196) | NGS Results | + | 399 | 101 | 500 | 98.2 (1817/1850); k = 0.714 (95% CI 0.620–0.807) (substantial) p = 0.1627 | 83.4 (399/478) | 98.5 (6673/6774) |
| − | 79 | 6673 | 6752 | |||||
| Total | 478 | 6774 | 7252 | |||||
| All epidemiological samples combined (n = 261) | NGS Results | + | 490 | 133 | 623 | 97.4 (9407/9657); k = 0.783 (95% CI 0.757–0.809) (substantial) p = 0.3472 | 80.7 (490/607) | 98.5 (8917/9050) |
| − | 117 | 8917 | 9034 | |||||
| Total | 607 | 9050 | 9657 | |||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lomsadze, A.; Li, T.; Rajeevan, M.S.; Unger, E.R.; Borodovsky, M. Bioinformatics Pipeline for Human Papillomavirus Short Read Genomic Sequences Classification Using Support Vector Machine. Viruses 2020, 12, 710. https://doi.org/10.3390/v12070710
Lomsadze A, Li T, Rajeevan MS, Unger ER, Borodovsky M. Bioinformatics Pipeline for Human Papillomavirus Short Read Genomic Sequences Classification Using Support Vector Machine. Viruses. 2020; 12(7):710. https://doi.org/10.3390/v12070710
Chicago/Turabian StyleLomsadze, Alexandre, Tengguo Li, Mangalathu S. Rajeevan, Elizabeth R. Unger, and Mark Borodovsky. 2020. "Bioinformatics Pipeline for Human Papillomavirus Short Read Genomic Sequences Classification Using Support Vector Machine" Viruses 12, no. 7: 710. https://doi.org/10.3390/v12070710
APA StyleLomsadze, A., Li, T., Rajeevan, M. S., Unger, E. R., & Borodovsky, M. (2020). Bioinformatics Pipeline for Human Papillomavirus Short Read Genomic Sequences Classification Using Support Vector Machine. Viruses, 12(7), 710. https://doi.org/10.3390/v12070710