Validation of Variant Assembly Using HAPHPIPE with Next-Generation Sequence Data from Viruses

 , , , and
, , , and 
Abstract
1. Introduction
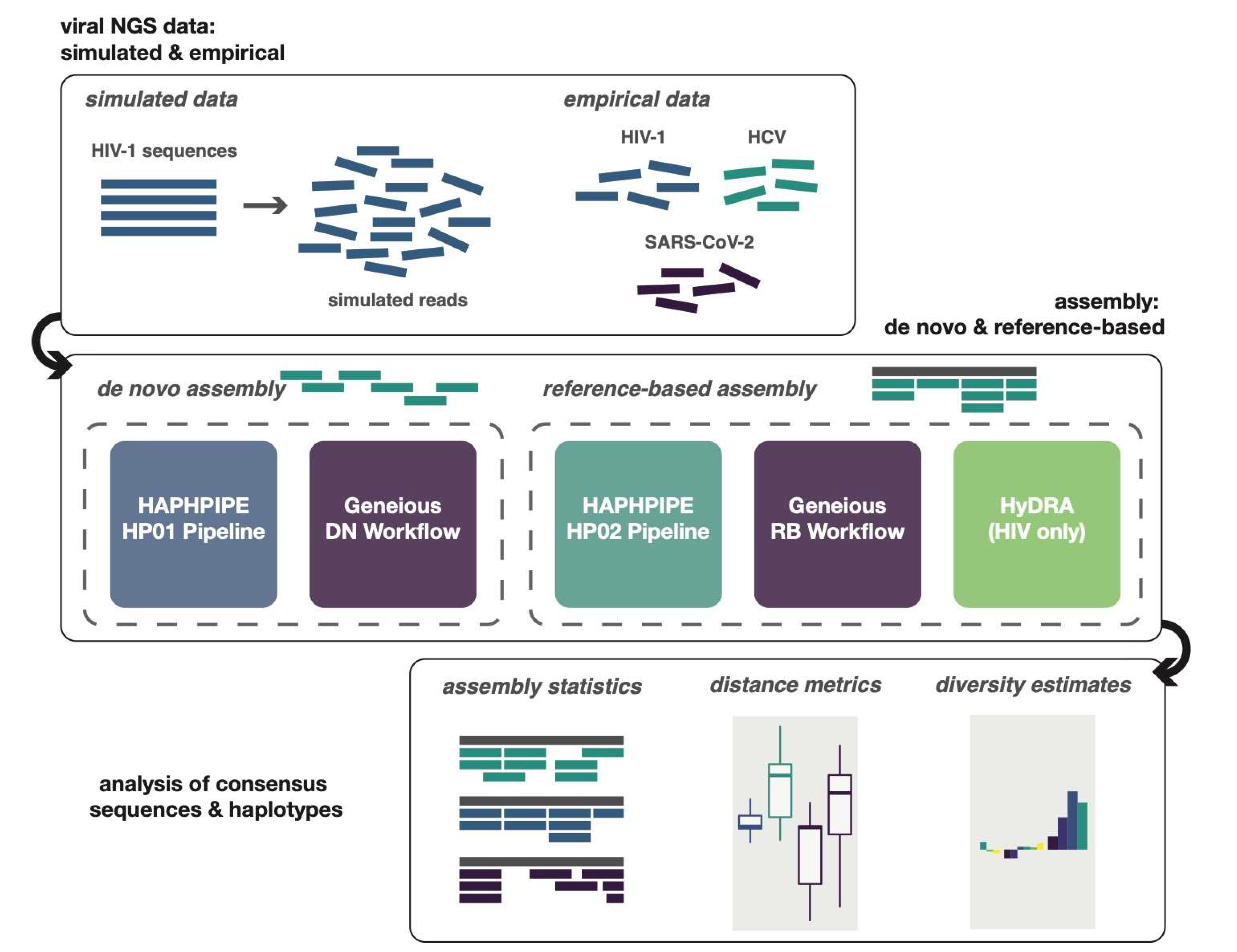
2. Materials and Methods
2.1. HAPHPIPE
2.2. HyDRA
2.3. Geneious
2.4. Data Simulation
2.5. Analyses and Testing
2.6. Empirical Data Applications
3. Results
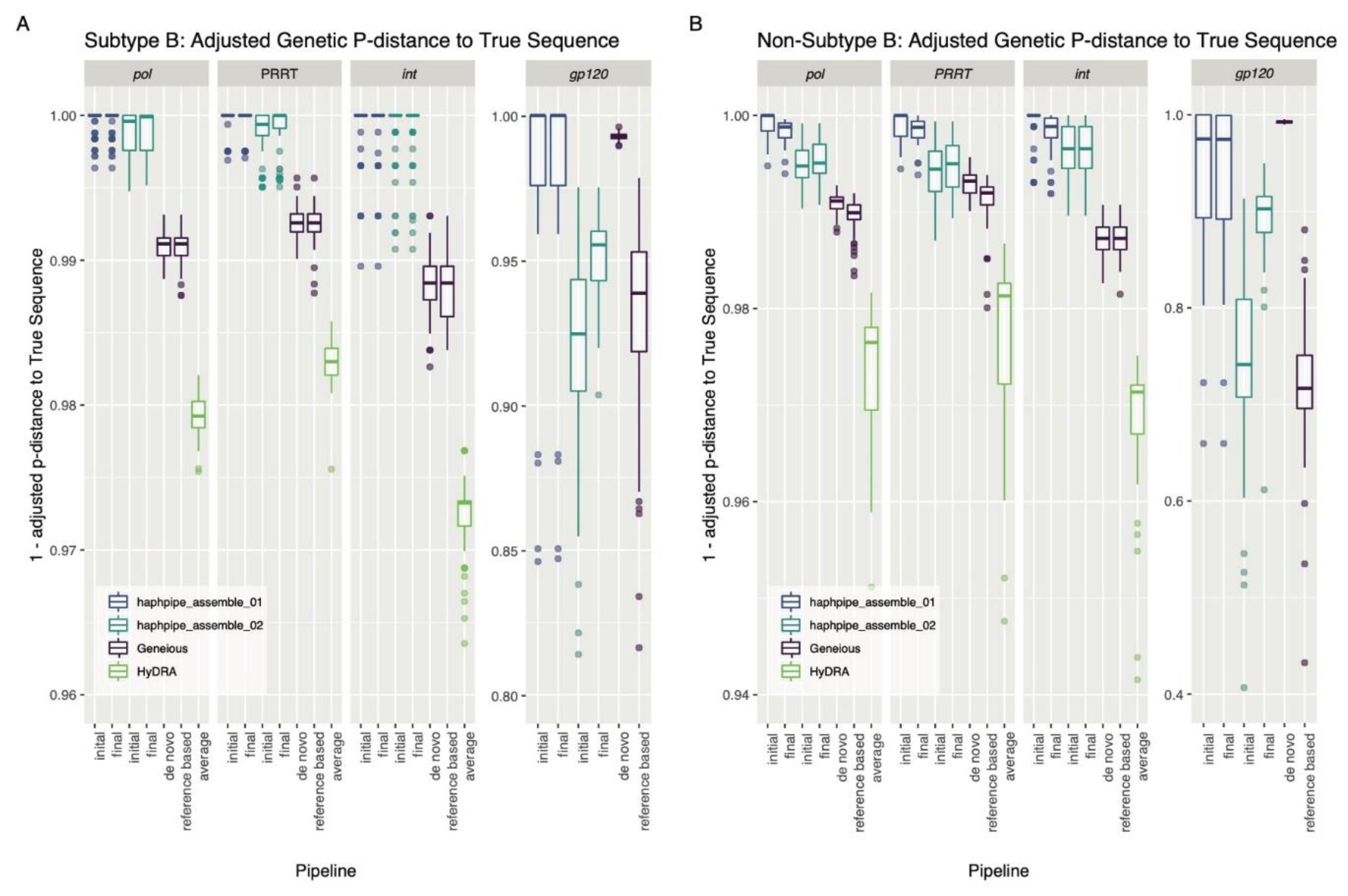
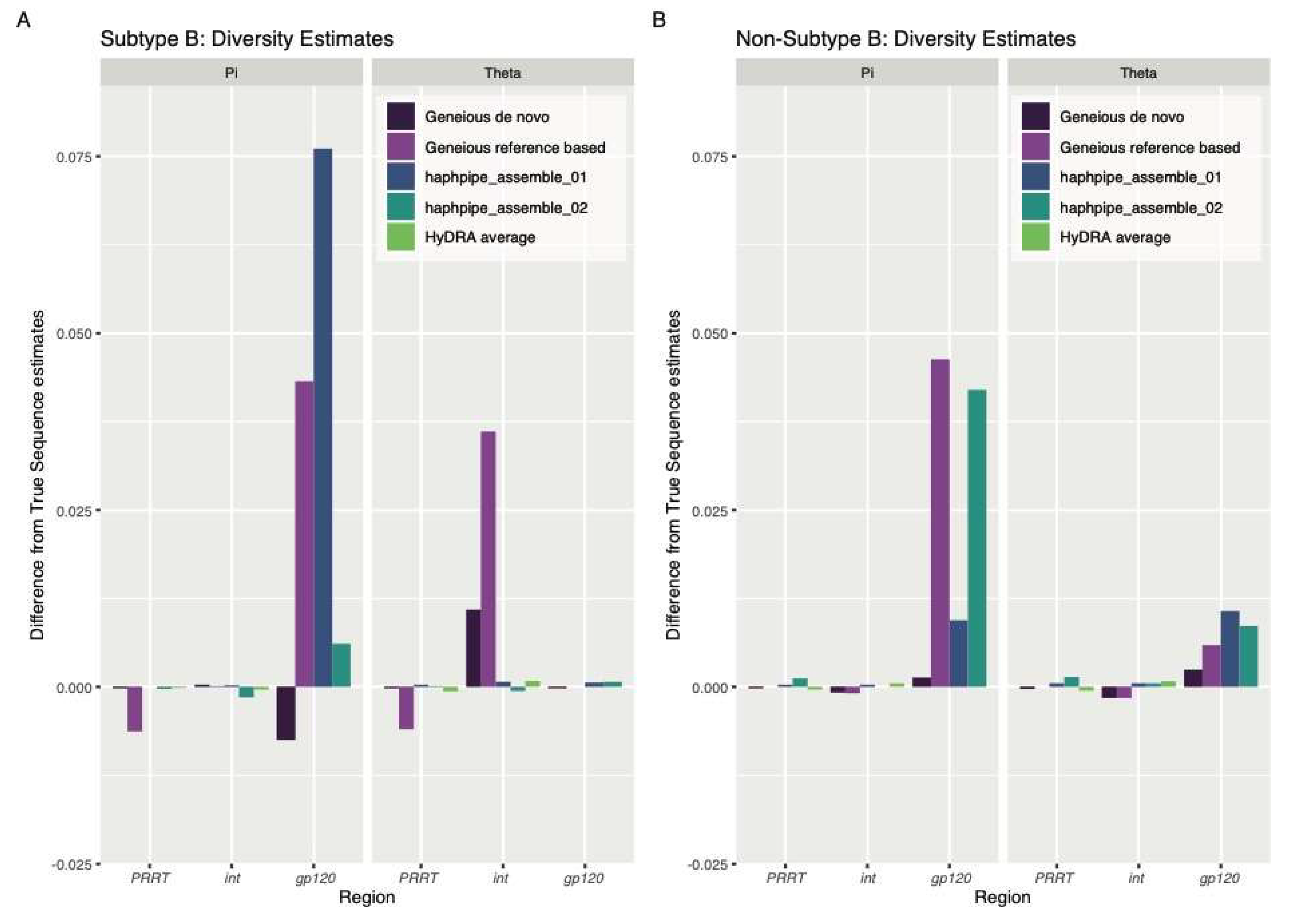
3.1. Simulated Data
3.2. Empirical Data
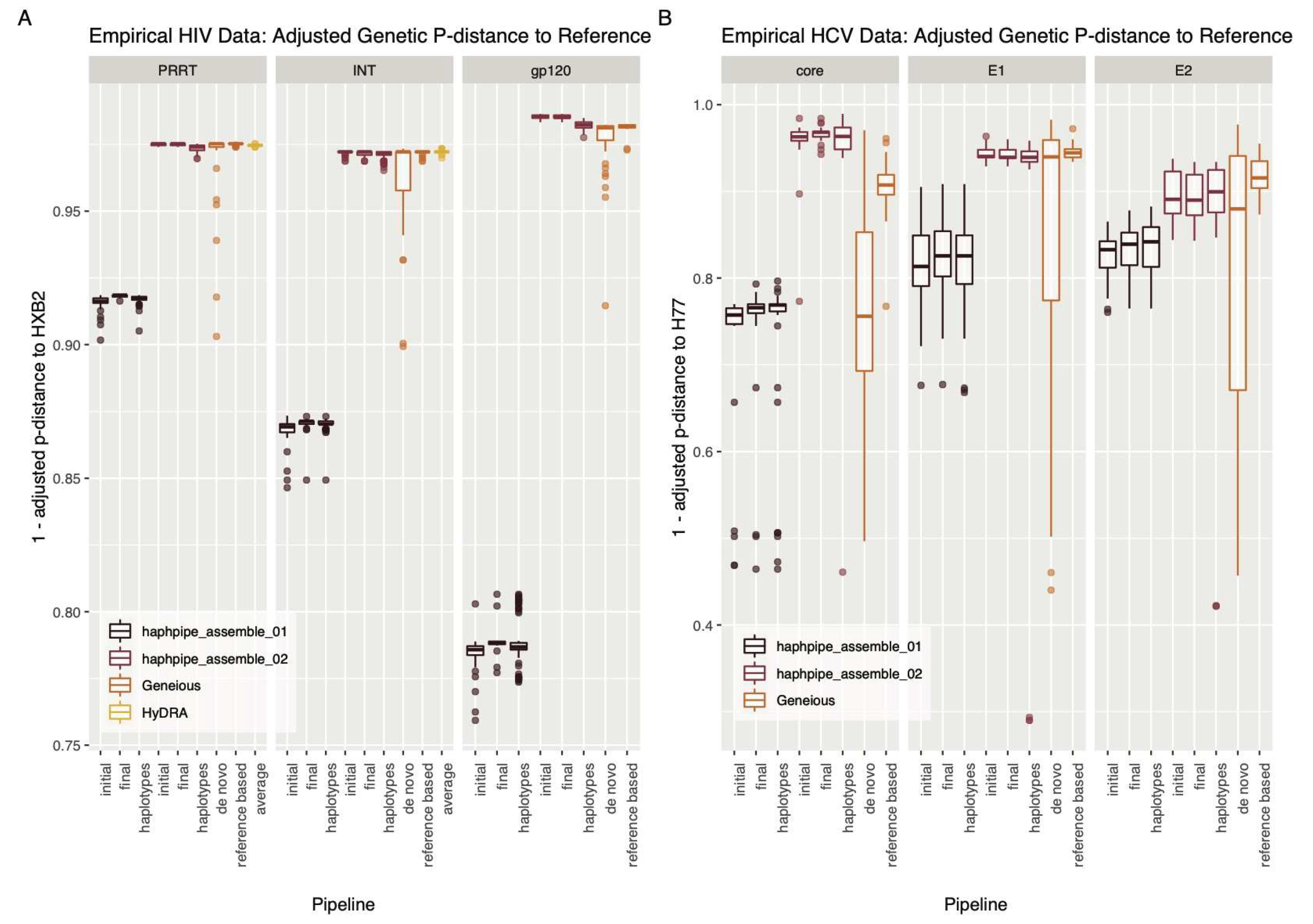
3.2.1. Empirical HIV Dataset
3.2.2. Empirical HCV Dataset
3.2.3. Empirical SARS-CoV-2 Dataset
3.2.4. Differences between Genetic Distance Measurements
4. Discussion
4.1. Simulated Data
4.2. Empirical Data
4.2.1. Assembly Statistics and Performance
4.2.2. HIV
4.2.3. HCV
4.2.4. SARS-CoV-2
4.2.5. Effects of Ambiguity Codes
4.3. Bias towards Reference Sequence
4.4. Limitations
4.5. Utility of Software
4.6. Comparison to other Viral Pipelines
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HP01 | HP02 | |||||||
|---|---|---|---|---|---|---|---|---|
| Sub B | Pseudomedian | CI Low | CI high | p Value | Pseudomedian | CI Low | CI High | p Value |
| pol | −0.0004 | NA 1 | NA 1 | 1 | 0.0004 | 0.0004 | 0.0004 | 2.3 × 10−06 *** |
| PRRT | −0.0004 | NA 1 | NA 1 | 0.3711 | 0.0006 | 0.0006 | 0.0006 | 3.6 × 10−06 *** |
| int | −0.0003 | NA 1 | NA 1 | 1 | 0.0009 | NA 1 | NA 1 | 1 |
| gp120 | 0.0008 | NA 1 | NA 1 | 0.3710 | 0.0298 | 0.0259 | 0.0333 | <2.2 × 10−16 *** |
| Non-B | Pseudomedian | CI Low | CI High | p Value | Pseudomedian | CI Low | CI high | p value |
| pol | −0.0008 | −0.0010 | −0.0008 | 2.5 × 10−09 *** | 0.0006 | 0.0005 | 0.0008 | 8.3 × 10−06 *** |
| PRRT | −0.0009 | −0.0010 | −0.0008 | 3.5 × 10−09 *** | 0.0009 | 0.0006 | 0.0011 | 8.4 × 10−06 *** |
| int | −0.0012 | −0.0017 | −0.0011 | 1.2 × 10−06 *** | 0.0017 | NA 1 | NA 1 | 1 |
| gp120 | −0.0007 | −0.0009 | −0.0006 | 4.7 × 10−06 *** | 0.1475 | 0.1296 | 0.1657 | 7.8 × 10−10 *** |
| Subtype B Simulation Data | Non-Subtype B Simulation Data | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Pipeline | HP01 | HP02 | GDN | GRB | HyDRA | HP01 | HP02 | GDN | GRB | HyDRA |
| pol | HP01 | 0.1168 *** | 1.1 × 10−25 *** | 3.4 × 10−27 *** | 2.7 × 10−73 *** | 0.0079 ** | 5.2 × 10−13 *** | 6.4 × 10−19 *** | 1.3 × 10−40 *** | ||
| HP02 | 0.1168 | 9.2 × 10−18 *** | 5.5 × 10−19 *** | 4.5 × 10−59 *** | 0.0079 ** | 1.7 × 10−05 *** | 2.7 × 10−09 *** | 1.9 × 10−25 *** | |||
| GDN | 1.1 × 10−25 *** | 9.2 × 10−18 *** | 0.7396 | 1.4 × 10−13 *** | 5.2 × 10−13 *** | 1.7 × 10−05 *** | 0.1021 | 7.9 × 10−09 *** | |||
| GRN | 3.4 × 10−27 *** | 5.5 × 10−19 *** | 0.7396 | 1.3 × 10−12 *** | 6.4 × 10−19 *** | 2.7 × 10−09 *** | 0.1021 | 3.2 × 10−05 *** | |||
| HyDRA | 2.7 × 10−73 *** | 4.5 × 10−59 *** | 1.4 × 10−13 *** | 1.3 × 10−12 *** | 1.3 × 10−40 *** | 1.9 × 10−25 *** | 7.9 × 10−09 *** | 3.2 × 10−05 *** | |||
| PRRT | HP01 | 0.1053 *** | 4.2 × 10−26 *** | 2.7 × 10−27 *** | 5.0 × 10−74 *** | 8.5 × 10−05 *** | 7.7 × 10−10 *** | 3.1 × 10−16 *** | 1.8 × 10−39 *** | ||
| HP02 | 0.1053 | 6.2 × 10−18 *** | 6.9 × 10−19 *** | 2.0 × 10−59 *** | 8.5 × 10−05 *** | 0.0499 * | 9.4 × 10−05 *** | 6.1 × 10−19 *** | |||
| GDN | 4.2 × 10−26 *** | 6.2 × 10−18 *** | 0.7918 | 1.4 × 10−13 *** | 7.7 × 10−10 *** | 0.0499 * | 0.0470 * | 3.9 × 10−11 *** | |||
| GRN | 2.7 × 10−27 *** | 6.9 × 10−19 *** | 0.7918 | 7.7 × 10−13 *** | 3.1 × 10−16 *** | 9.4 × 10−05 *** | 0.0470 * | 4.8 × 10−06 *** | |||
| HyDRA | 5.0 × 10−74 *** | 2.0 × 10−59 *** | 1.4 × 10−13 *** | 7.7 × 10−13 *** | 1.8 × 10−39 *** | 6.1 × 10−19 *** | 3.9 × 10−11 *** | 4.8 × 10−06 *** | |||
| int | HP01 | 1 | 8.7 × 10−23 *** | 1.1 × 10−23 *** | 9.4 × 10−69 *** | 0.5092 | 2.0 × 10−12 *** | 1.2 × 10−13 *** | 6.0 × 10−36 *** | ||
| HP02 | 1 | 1.4 × 10−21 *** | 2.0 × 10−22 *** | 2.1 × 10−66 *** | 0.5092 | 3.7 × 10−09 *** | 3.5 × 10−10 *** | 5.9 × 10−30 *** | |||
| GDN | 8.7 × 10−23 *** | 1.4 × 10−21 *** | 0.8290 | 9.1 × 10−14 *** | 2.0 × 10−12 *** | 3.7 × 10−09 *** | 0.6963 | 2.7 × 10−07 *** | |||
| GRN | 1.1 × 10−23 *** | 2.0 × 10−22 *** | 0.8290 | 3.6 × 10−13 *** | 1.2 × 10−13 *** | 3.5 × 10−10 *** | 0.6963 | 1.6 × 10−06 *** | |||
| HyDRA | 9.4 × 10−69 *** | 2.1 × 10−66 *** | 9.1 × 10−14 *** | 3.6 × 10−13 *** | 6.0 × 10−36 *** | 5.9 × 10−30 *** | 2.7 × 10−07 *** | 1.6 × 10−06 *** | |||
| gp120 | HP01 | 2.8 × 10−26 *** | 0.4269 | 2.0 × 10−40 *** | NA | 6.5 × 10−05 *** | 0.1212 | 2.5 × 10−19 *** | NA | ||
| HP02 | 2.8 × 10−26 *** | 8.3 × 10−23 *** | 0.0134 * | NA | 6.5 × 10−05 *** | 4.7 × 10−08 *** | 1.6 × 10−06 *** | NA | |||
| GDN | 0.4269 | 8.3 × 10−23 *** | 5.5 × 10−36 *** | NA | 0.1212 | 4.7 × 10−08 *** | 5.3 × 10−26 *** | NA | |||
| GRN | 2.0 × 10−40 *** | 0.0134 * | 5.5 × 10−36 *** | NA | 2.5 × 10−19 *** | 1.6 × 10−06 *** | 5.3 × 10−26 *** | NA | |||
| HP01 | HP02 | |||||||
|---|---|---|---|---|---|---|---|---|
| Sub B | pseudomedian | CI low | CI high | p value | pseudomedian | CI low | CI high | p value |
| pol | 0.0008 | NA 1 | NA 1 | 1 | 8.9 × 10−06 | −7.2 × 10−05 | 0.0001 | 0.2074 |
| PRRT | 0.0002 | NA 1 | NA 1 | 1 | −2.6 × 10−05 | −0.0002 | 0.0002 | 0.1670 |
| int | 0.0020 | NA 1 | NA 1 | 1 | 0.0009 | NA 1 | NA 1 | 1 |
| gp120 | 0.0006 | 0.0006 | 0.0006 | 0.3711 | 0.0135 | 0.0105 * | 0.0169 | 2.1 × 10−13 *** |
| Non-B | pseudomedian | CI low | CI high | p value | pseudomedian | CI low | CI high | p value |
| pol | −0.0009 | −0.0010 | −0.0008 | 1.8 × 10−09 *** | 0.0001 | −2.8 × 10−05 | 0.0003 | 0.1692 |
| PRRT | −0.0009 | −0.0010 | −0.0008 | 4.6 × 10−09 *** | 0.0002 | −3.7 × 10−05 | 0.0004 | 0.1614 |
| int | −0.0012 | −0.0017 | −0.0012 | 2.1 × 10−07 *** | −0.0006 | NA 1 | NA 1 | 1 |
| gp120 | −0.0006 | −0.0007 | −0.0005 | 5.9 × 10−05 *** | 0.1070 | 0.0949 | 0.1209 | 7.8 × 10−10 *** |
| Subtype B Simulation Data | Non-Subtype B Simulation Data | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Pipeline | HP01 | HP02 | GDN | GRB | HyDRA | HP01 | HP02 | GDN | GRB | HyDRA |
| pol | HP01 | 1 | 2.3 × 10−08 *** | 1.4 × 10−08 *** | 2.7 × 10−38 *** | 0.8790 | 0.0013 ** | 0.0002 *** | 1.2 × 10−17 *** | ||
| HP02 | 1 | 5.8 × 10−08 *** | 4.0 × 10−08 *** | 5.6 × 10−37 *** | 0.8790 | 0.0120 * | 0.0029 *** | 7.9 × 10−15 *** | |||
| GDN | 2.3 × 10−08 *** | 5.8 × 10−08 *** | 0.9104 | 3.2 × 10−12 *** | 0.0013 | 0.0120 * | 0.6142 *** | 1.9 × 10−06 *** | |||
| GRN | 1.4 × 10−08 *** | 4.0 × 10−08 *** | 0.9104 | 6.3 × 10−12 *** | 0.0002 *** | 0.0029 *** | 0.6142 | 2.2 × 10−05 *** | |||
| HyDRA | 2.7 × 10−38 *** | 5.6 × 10−37 *** | 3.2 × 10−12 *** | 6.3 × 10−12 *** | 1.2 × 10−17 *** | 7.9 × 10−15 *** | 1.9 × 10−06 *** | 2.2 × 10−05 *** | |||
| PRRT | HP01 | 1 | 0.0001 *** | 7.8 × 10−05 *** | 5.9 × 10−24 *** | 0.7740 | 0.0387 * | 0.0099 ** | 6.8 × 10−13 *** | ||
| HP02 | 1 | 0.0004 *** | 0.0003 *** | 2.9 × 10−22 *** | 0.7740 | 0.2163 | 0.0897 | 3.1 × 10−10 *** | |||
| GDN | 0.0001 *** | 0.0004 *** | 0.9049 | 9.8 × 10−09 *** | 0.0387 * | 0.2163 | 0.6279 | 1.1 × 10−05 *** | |||
| GRN | 7.8 × 10−05 *** | 0.0003 *** | 0.9049 | 1.8 × 10−08 *** | 0.0099 ** | 0.0897 | 0.6279 | 9.8 × 10−05 *** | |||
| HyDRA | 5.9 × 10−24 *** | 2.9 × 10−22 *** | 9.8 × 10−09 *** | 1.80 × 10−08 *** | 6.8 × 10−13 *** | 3.1 × 10−10 *** | 1.1 × 10−05 *** | 9.8 × 10−05 *** | |||
| int | HP01 | 0.9649 | 6.0 × 10−12 *** | 4.7 × 10−12 *** | 7.5 × 10−49 *** | 1 | 0.0002 *** | 0.0001 *** | 3.2 × 10−19 *** | ||
| HP02 | 0.9649 | 5.8 × 10−12 *** | 4.1 × 10−12 *** | 4.3 × 10−49 *** | 1 | 0.0005 *** | 0.0003 *** | 5.0 × 10−18 *** | |||
| GDN | 6.0 × 10−12 *** | 5.8 × 10−12 *** | 1 | 4.9 × 10−14 *** | 0.0002 *** | 0.0005 *** | 0.8497 | 2.4 × 10−06 *** | |||
| GRN | 4.7 × 10−12 *** | 4.1 × 10−12 *** | 1 | 9.6 × 10−14 *** | 0.0001 *** | 0.0003 *** | 0.8497 | 5.6 × 10−06 *** | |||
| HyDRA | 7.5 × 10−49 *** | 4.3 × 10−49 *** | 4.9 × 10−14 *** | 9.6 × 10−14 *** | 3.2 × 10−19 *** | 5.0 × 10−18 *** | 2.4 × 10−06 *** | 5.6 × 10−06 *** | |||
| gp120 | HP01 | NA 1 | NA 1 | NA 1 | NA 1 | 0.1905 | 0.1731 | 3.4 × 10−11 *** | NA | ||
| HP02 | NA 1 | NA 1 | NA 1 | NA 1 | 0.1905 | 0.8873 | 2.2 × 10−17 *** | NA | |||
| GDN | NA 1 | NA 1 | NA 1 | NA 1 | 0.1731 | 0.8873 | 6.4 × 10−17 *** | NA | |||
| GRN | NA 1 | NA 1 | NA 1 | NA 1 | 3.4 × 10−11 *** | 2.2 × 10−17 *** | 6.4 × 10−17 *** | NA | |||
| Simulated Data Subtype B | |||||||||||||
| PRRT | int | gp120 | |||||||||||
| Pipeline | N | H | S | π | θ | H | S | π | θ | H | S | π | θ |
| True sequences | 100 | 100 | 826 | 0.0654 | 0.0987 | 100 | 389 | 0.0510 | 0.0870 | 100 | 1407 | 0.2219 | 0.1913 |
| HP01 | 100 | 100 | 824 | 0.0654 | 0.0984 | 100 | 386 | 0.0508 | 0.0863 | 100 | 1378 | 0.1458 | 0.1831 |
| HP02 | 100 | 100 | 823 | 0.0657 | 0.0986 | 100 | 388 | 0.0525 | 0.0876 | 100 | 1362 | 0.2158 | 0.1877 |
| HyDRA | 100 | 100 | 820 | 0.0656 | 0.0994 | 100 | 377 | 0.0514 | 0.0862 | NA | NA | NA | NA |
| GRB | 100 | 100 | 835 | 0.0717 | 0.1047 | 100 | 384 | 0.0509 | 0.0509 | 100 | 1234 | 0.1787 | 0.1657 |
| GDN | 100 | 100 | 823 | 0.0656 | 0.0989 | 100 | 384 | 0.0507 | 0.0761 | 100 | 1421 | 0.2294 | 0.1930 |
| Simulated Data non-Subtype B | |||||||||||||
| Pipeline | N | H | S | π | θ | H | S | π | θ | H | S | π | θ |
| True sequences | 50 | 50 | 753 | 0.1016 | 0.1039 | 50 | 339 | 0.0749 | 0.0876 | 50 | 1185 | 0.2419 | 0.1809 |
| HP01 | 50 | 50 | 749 | 0.1013 | 0.1034 | 50 | 337 | 0.0746 | 0.0871 | 50 | 1106 | 0.2325 | 0.1702 |
| HP02 | 50 | 50 | 746 | 0.1004 | 0.1025 | 50 | 335 | 0.0749 | 0.0871 | 50 | 957 | 0.1999 | 0.1723 |
| HyDRA | 50 | 50 | 745 | 0.1020 | 0.1045 | 50 | 328 | 0.0749 | 0.0868 | NA | NA | NA | NA |
| GRB | 50 | 50 | 748 | 0.1016 | 0.1039 | 50 | 338 | 0.0758 | 0.0892 | 50 | 916 | 0.1956 | 0.1750 |
| GDN | 50 | 50 | 751 | 0.1018 | 0.1042 | 50 | 338 | 0.0757 | 0.0892 | 50 | 1180 | 0.2406 | 0.1785 |
| HP01 | HP02 | |||||||
|---|---|---|---|---|---|---|---|---|
| Empirical HIV dataset | ||||||||
| pseudomedian | CI low | CI high | p value | pseudomedian | CI low | CI high | p value | |
| PRRT | 0.0022 | 0.0017 | 0.0035 | 1.5 × 10−06 *** | NA 1 | NA 1 | NA 1 | NA 1 |
| int | 0.0021 | 0.0016 | 0.0031 | 2.2 × 10−05 *** | −0.0012 | NA 1 | NA 1 | 0.1489 |
| gp120 | 0.0026 | 0.0020 | 0.0038 | 2.6 × 10−07 *** | −0.0005 | −0.0006 | −0.0005 | 0.0029 ** |
| Empirical HCV dataset | ||||||||
| pseudomedian | CI low | CI high | p value | pseudomedian | CI low | CI high | p value | |
| core | 0.0083 | 0.0021 | 0.0125 | 0.0012 ** | 0.0101 | 0.0025 | 0.1027 | 0.0412 * |
| E1 | 0.0086 | 0.0054 | 0.0121 | 0.0001 *** | −0.0026 | −0.0069 | −0.0017 | 0.0579 |
| E2 | 0.0079 | 0.0048 | 0.0144 | 0.0001 *** | −0.0027 | −0.0040 | −0.0013 | 0.0001 *** |
| Gene | Pipeline | HP01 | HP02 | HP01 Haps | HP02 Haps | GDN | GRB | HyDRA |
|---|---|---|---|---|---|---|---|---|
| Pol | HP01 | 4.6 × 10−14 *** | 0.4447 | 1.0 × 10−09 *** | 2.1 × 10−10 *** | 2.0 × 10−15 *** | 1.5 × 10−08 *** | |
| HP02 | 4.6 × 10−14 *** | 3.2 × 10−30 *** | 0.0058 ** | 0.7591 | 0.6986 | 0.4900 | ||
| HP01 haps | 0.4447 | 3.2 × 10−30 *** | 5.1 × 10−36 *** | 2.3 × 10−23 *** | 1.0 × 10−32 *** | 8.9 × 10−20 *** | ||
| HP02 haps | 1.0 × 10−09 *** | 0.0058 ** | 5.1 × 10−36 *** | 0.3641 | 0.0009 *** | 0.9894 | ||
| GDN | 2.1 × 10−10 *** | 0.7591 | 2.3 × 10−23 *** | 0.3641 | 0.6298 | 1 | ||
| GRB | 2.0 × 10−15 *** | 0.6986 | 1.0 × 10−32 *** | 0.0009 *** | 0.6298 | 0.2508 | ||
| HyDRA | 1.5 × 10−08 *** | 0.4900 | 8.9 × 10−20 *** | 0.9894 | 1 | 0.2508 | ||
| Int | HP01 | 4.9 × 10−12 *** | 1 | 1.6 × 10−14 *** | 2.4 × 10−09 *** | 2.2 × 10−13 *** | 6.0 × 10−16 *** | |
| HP02 | 4.9 × 10−12 *** | 1.0 × 10−23 *** | 1 | 1 | 1 | 1 | ||
| HP01 haps | 1 | 1.0 × 10−23 *** | 3.7 × 10−47 *** | 8.1 × 10−19 *** | 3.5 × 10−26 *** | 7.7 × 10−31 *** | ||
| HP02 haps | 1.6 × 10−14 *** | 1 | 3.7 × 10−47 *** | 0.9955 | 0.8553 | 0.1024 | ||
| GDN | 2.4 × 10−09 *** | 1 | 8.1 × 10−19 *** | 0.9955 | 1 | 0.3966 | ||
| GRB | 2.2 × 10−13 *** | 1 | 3.5 × 10−26 *** | 0.8553 | 1 | 1 | ||
| HyDRA | 6.0 × 10−16 *** | 1 | 7.7 × 10−31 *** | 0.1024 | 0.3966 | 1 | ||
| gp120 | HP01 | 1.8 × 10−14 *** | 0.0455 * | 2.6 × 10−09 *** | 0.0001 *** | 3.2 × 10−06 *** | NA | |
| HP02 | 1.8 × 10−14 *** | 2.1 × 10−37 *** | 0.0228 * | 0.0023 ** | 0.0241 * | NA | ||
| HP01 haps | 0.0455 * | 2.1 × 10−37 *** | 2.8 × 10−36 *** | 1.5 × 10−15 *** | 4.1 × 10−19 *** | NA | ||
| HP02 haps | 2.6 × 10−09 *** | 0.0228 * | 2.8 × 10−36 *** | 0.5908 | 0.6382 | NA | ||
| GDN | 0.0001 *** | 0.0023 * | 1.5 × 10−15 *** | 0.5908 | 0.9509 | NA | ||
| GRB | 3.2 × 10−06 *** | 0.0241 * | 4.1 × 10−19 *** | 0.6382 | 0.9509 | NA |
| Gene | Pipeline | HP01 | HP02 | HP01 Haps | HP02 Haps | GDN | GRB | HyDRA |
|---|---|---|---|---|---|---|---|---|
| Core | HP01 | 1.0 × 10−10 *** | 0.7016 | 6.8 × 10−14 *** | 1 | 0.0027 ** | ||
| HP02 | 1.0 × 10−10 *** | 4.6 × 10−14 *** | 1 | 3.0 × 10−08 *** | 0.0071 ** | 1.0 × 10−10 *** | ||
| HP01 haps | 0.7016 | 4.6 × 10−14 *** | 2.0 × 10−23 *** | 1 | 0.0007 *** | 0.7016 | ||
| HP02 haps | 6.8 × 10−14 *** | 1 | 2.0 × 10−23 *** | 1.9 × 10−10 *** | 0.0039 ** | 6.8 × 10−14 *** | ||
| GDN | 1 | 3.0 × 10−08 *** | 1 | 1.9 × 10−10 *** | 0.0359 * | 1 | ||
| GRB | 0.0027 ** | 0.0071 ** | 0.0007 *** | 0.0039 ** | 0.0359 * | 0.0027 ** | ||
| E1 | HP01 | 9.0 × 10−07 *** | 0.9367 | 1.0 × 10−06 *** | 0.0014 ** | 3.5 × 10−08 *** | ||
| HP02 | 9.0 × 10−07 *** | 3.5 × 10−09 *** | 1 | 0.6392 | 1 | 9.0 × 10−07 *** | ||
| HP01 haps | 0.9367 | 3.5 × 10−09 *** | 2.4 × 10−10 *** | 8.2 × 10−05 *** | 4.3 × 10−11 *** | 0.9367 | ||
| HP02 haps | 1.0 × 10−06 *** | 1 | 2.4 × 10−10 *** | 1 | 0.5477 | 1.0 × 10−06 *** | ||
| GDN | 0.0014 ** | 0.6392 | 8.2 × 10−05 *** | 1 | 0.2005 | 0.0014 ** | ||
| GRB | 3.5 × 10−08 *** | 1 | 4.3 × 10−11 *** | 0.5477 | 0.2005 | 3.5 × 10−08 *** | ||
| E2 | HP01 | 2.8 × 10−04 *** | 1 | 3.6 × 10−06 *** | 0.0685 | 5.5 × 10−08 *** | ||
| HP02 | 2.8 × 10−04 *** | 1.7 × 10−05 *** | 0.8929 | 0.3236 | 0.3692 | 2.8 × 10−04 *** | ||
| HP01 haps | 1 | 1.7 × 10−05 *** | 8.7 × 10−10 *** | 0.0327 * | 1.2 × 10−10 *** | 1 | ||
| HP02 haps | 3.6 × 10−06 *** | 0.8929 | 8.7 × 10−10 *** | 0.2427 | 0.3076 | 3.6 × 10−06 *** | ||
| GDN | 0.0685 | 0.3236 | 0.0327 * | 0.2427 | 0.0090 ** | 0.0685 | ||
| GRB | 5.5 × 10−08 *** | 0.3692 | 1.2 × 10−10 *** | 0.3076 | 0.0090 ** | 5.5 × 10−08 *** |
References
- Zanini, F.; Brodin, J.; Thebo, L.; Lanz, C.; Bratt, G.; Albert, J.; Neher, R.A. Population genomics of intrapatient HIV-1 evolution. Elife 2015, 4, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Bonnaud, E.M.; Troupin, C.; Dacheux, L.; Holmes, E.C.; Monchatre-Leroy, E.; Tanguy, M.; Bouchier, C.; Cliquet, F.; Barrat, J.; Bourhy, H. Comparison of intra- and inter-host genetic diversity in rabies virus during experimental cross-species transmission. PLOS Pathog. 2019, 15, e1007799. [Google Scholar] [CrossRef]
- Pagán, I. The diversity, evolution and epidemiology of plant viruses: A phylogenetic view. Infect. Genet. Evol. 2018, 65, 187–199. [Google Scholar] [CrossRef]
- Pérez-Losada, M.; Arenas, M.; Galán, J.C.; Bracho, M.A.; Hillung, J.; García-González, N.; González-Candelas, F. High-throughput sequencing (HTS) for the analysis of viral populations. Infect. Genet. Evol. 2020, 80, 104208. [Google Scholar] [CrossRef] [PubMed]
- Simpson, J.T.; Pop, M. The theory and practice of genome sequence assembly. Annu. Rev. Genom. Hum. Genet. 2015, 16, 153–172. [Google Scholar] [CrossRef] [PubMed]
- Nagarajan, N.; Pop, M. Sequence assembly demystified. Nat. Rev. Genet. 2013, 14, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Günther, T.; Nettelblad, C. The presence and impact of reference bias on population genomic studies of prehistoric human populations. PLoS Genet. 2019, 15, 1–20. [Google Scholar] [CrossRef]
- Posada-Cespedes, S.; Seifert, D.; Beerenwinkel, N. Recent advances in inferring viral diversity from high-throughput sequencing data. Virus Res. 2017, 239, 17–32. [Google Scholar] [CrossRef]
- Archer, J.; Rambaut, A.; Taillon, B.E.; Richard Harrigan, P.; Lewis, M.; Robertson, D.L. The evolutionary analysis of emerging low frequency HIV-1 CXCR4 using variants through time-an ultra-deep approach. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef]
- Ji, H.; Enns, E.; Brumme, C.J.; Parkin, N.; Howison, M.; Lee, E.R.; Capina, R.; Marinier, E.; Avila-Rios, S.; Sandstrom, P.; et al. Bioinformatic data processing pipelines in support of next-generation sequencing-based HIV drug resistance testing: The Winnipeg Consensus. J. Int. AIDS Soc. 2018, 21, 1–14. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Korber, B.; Foley, B.T.; Kuiken, C.; Pillai, S.K.; Sodroski, J.G. Others Numbering positions in HIV relative to HXB2CG. AIDS Res. Hum. Retrovir. 1998, 3, 102–111. [Google Scholar]
- Banin, A.N.; Tuen, M.; Bimela, J.S.; Tongo, M.; Zappile, P.; Khodadadi-Jamayran, A.; Nanfack, A.J.; Meli, J.; Wang, X.; Mbanya, D.; et al. Development of a versatile, near full genome amplification and sequencing approach for a broad variety of HIV-1 group M variants. Viruses 2019, 11, 317. [Google Scholar] [CrossRef]
- Kijak, G.H.; Sanders-Buell, E.; Pham, P.; Harbolick, E.A.; Oropeza, C.; O’Sullivan, A.M.; Bose, M.; Beckett, C.G.; Milazzo, M.; Robb, M.L.; et al. Next-generation sequencing of HIV-1 single genome amplicons. Biomol. Detect. Quantif. 2019, 17, 100080. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Olivo, A.; Laeyendecker, O.; Forberg, K.; Ndembi, N.; Mbanya, D.; Kaptue, L.; Quinn, T.C.; Cloherty, G.A.; Rodgers, M.A.; et al. Universal target capture of HIV sequences from NGS libraries. Front. Microbiol. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Berg, M.G.; Yamaguchi, J.; Alessandri-Gradt, E.; Tell, R.W.; Plantier, J.C.; Brennan, C.A. A pan-HIV strategy for complete genome sequencing. J. Clin. Microbiol. 2016, 54, 868–882. [Google Scholar] [CrossRef]
- Fonager, J.; Larsson, J.T.; Hussing, C.; Engsig, F.N.; Nielsen, C.; Fischer, T.K. Identification of minority resistance mutations in the HIV-1 integrase coding region using next generation sequencing. J. Clin. Virol. 2015, 73, 95–100. [Google Scholar] [CrossRef]
- Pessoa, R.; Sanabani, S.S. High prevalence of HIV-1 transmitted drug- resistance mutations from proviral DNA massively parallel sequencing data of therapy- na ï ve chronically infected Brazilian blood donors. PLoS ONE 2017, 12, e0785559. [Google Scholar] [CrossRef]
- Dudley, D.M.; Bailey, A.L.; Mehta, S.H.; Hughes, A.L.; Kirk, G.D.; Westergaard, R.P.; O’Connor, D.H. Cross-clade simultaneous HIV drug resistance genotyping for reverse transcriptase, protease, and integrase inhibitor mutations by Illumina MiSeq. Retrovirology 2014, 11, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Matume, N.D.; Tebit, D.M.; Gray, L.R.; Hammarskjold, M.L.; Rekosh, D.; Bessong, P.O. Next generation sequencing reveals a high frequency of CXCR4 utilizing viruses in HIV-1 chronically infected drug experienced individuals in South Africa. J. Clin. Virol. 2018, 103, 81–87. [Google Scholar] [CrossRef] [PubMed]
- Perrier, M.; Désiré, N.; Storto, A.; Todesco, E.; Rodriguez, C.; Bertine, M.; Le Hingrat, Q.; Visseaux, B.; Calvez, V.; Descamps, D.; et al. Evaluation of different analysis pipelines for the detection of HIV-1 minority resistant variants. PLoS ONE 2018, 13, 0198334. [Google Scholar] [CrossRef]
- Alves, B.M.; Siqueira, J.D.; Prellwitz, I.M.; Botelho, O.M.; Da Hora, V.P.; Sanabani, S.; Recordon-Pinson, P.; Fleury, H.; Soares, E.A.; Soares, M.A. Estimating HIV-1 genetic diversity in Brazil through next-generation sequencing. Front. Microbiol. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Alves, B.M.; Siqueira, J.D.; Garrido, M.M.; Botelho, O.M.; Prellwitz, I.M.; Ribeiro, S.R.; Soares, E.A.; Soares, M.A. Genome Quasispecies from Patients with Undetectable Viral Load Undergoing First-Line HAART Therapy. Viruses 2017, 9, 392. [Google Scholar] [CrossRef] [PubMed]
- Derache, A.; Iwuji, C.C.; Danaviah, S.; Giandhari, J.; Marcelin, A.-G.; Calvez, V.; de Oliveira, T.; Dabis, F.; Pillay, D.; Gupta, R.K. Predicted antiviral activity of tenofovir versus abacavir in combination with a cytosine analogue and the integrase inhibitor dolutegravir in HIV-1-infected South African patients initiating or failing first-line ART. J. Anitmicrobial Chemother. 2019, 74, 473–479. [Google Scholar] [CrossRef] [PubMed]
- Ávila-Ríos, S.; García-Morales, C.; Matías-Florentino, M.; Romero-Mora, K.A.; Tapia-Trejo, D.; Quiroz-Morales, V.S.; Reyes-Gopar, H.; Ji, H.; Sandstrom, P.; Casillas-Rodríguez, J.; et al. Pretreatment HIV-drug resistance in Mexico and its impact on the effectiveness of first-line antiretroviral therapy: A nationally representative 2015 WHO survey. Lancet HIV 2016, 3, e579–e591. [Google Scholar] [CrossRef]
- Taylor, T.; Lee, E.R.; Nykoluk, M.; Enns, E.; Liang, B.; Capina, R.; Gauthier, M.K.; Van Domselaar, G.; Sandstrom, P.; Brooks, J.; et al. A MiSeq-HyDRA platform for enhanced HIV drug resistance genotyping and surveillance. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef]
- Arias, A.; López, P.; Sánchez, R.; Yamamura, Y.; Rivera-Amill, V. Sanger and next generation sequencing approaches to evaluate HIV-1 virus in blood compartments. Int. J. Environ. Res. Public Health 2018, 15, 1697. [Google Scholar] [CrossRef]
- Hora, B.; Keating, S.M.; Chen, Y.; Sanchez, A.M.; Sabino, E.; Hunt, G.; Ledwaba, J.; Hackett, J.; Swanson, P.; Hewlett, I.; et al. Genetic characterization of a panel of diverse HIV-1 isolates at seven international sites. PLoS ONE 2016, 11, 0157340. [Google Scholar] [CrossRef]
- Nguyen, T.; Fofana, D.B.; Lê, M.P.; Charpentier, C.; Peytavin, G.; Wirden, M.; Lambert-Niclot, S.; Desire, N.; Grude, M.; Morand-Joubert, L.; et al. Prevalence and clinical impact of minority resistant variants in patients failing an integrase inhibitor-based regimen by ultra-deep sequencing. J. Antimicrob. Chemother. 2018, 73, 2485–2492. [Google Scholar] [CrossRef]
- Charpentier, C.; Montes, B.; Perrier, M.; Meftah, N.; Reynes, J. HIV-1 DNA ultra-deep sequencing analysis at initiation of the dual therapy dolutegravir ! lamivudine in the maintenance DOLULAM pilot study. J. Anitmicrobial Chemother. 2017, 72, 2831–2836. [Google Scholar] [CrossRef]
- Inzaule, S.C.; Hamers, R.L.; Noguera-julian, M.; Casadella, M.; Parera, M.; De Wit, T.F.R.; Paredes, R. Primary resistance to integrase strand transfer inhibitors in patients infected with diverse HIV-1 subtypes in sub-Saharan Africa. J. Anitmicrobial Chemother. 2018, 73, 1167–1172. [Google Scholar] [CrossRef] [PubMed]
- Dalmat, R.R.; Makhsous, N.; Pepper, G.G.; Magaret, A.; Jerome, K.R.; Wald, A. Limited Marginal Utility of Deep Sequencing for HIV Drug Resistance Testing in the Age of Integrase Inhibitors. J. Clin. Microbiol. 2018, 56, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Redd, A.D.; Mullis, C.E.; Serwadda, D.; Kong, X.; Martens, C.; Ricklefs, S.M.; Tobian, A.A.R.; Xiao, C.; Grabowski, M.K.; Nalugoda, F.; et al. The Rates of HIV Superinfection and Primary HIV Incidence in a General Population in Rakai, Uganda. J. Infect. Dis. 2012, 206, 267–274. [Google Scholar] [CrossRef]
- Eshleman, S.H.; Hudelson, S.E.; Redd, A.D.; Wang, L.; Debes, R.; Chen, Y.Q.; Martens, C.A.; Ricklefs, S.M.; Selig, E.J.; Porcella, S.F.; et al. Analysis of genetic linkage of HIV from couples enrolled in the HIV Prevention Trials Network 052 trial. J. Infect. Dis 2011, 204, 1918–1926. [Google Scholar] [CrossRef] [PubMed]
- Tumiotto, C.; Riviere, L.; Bellecave, P.; Recordon-pinson, P.; Latitude, P.; Vilain-parce, A.; Guidicelli, G. Sanger and Next-Generation Sequencing data for characterization of CTL epitopes in archived HIV-1 proviral DNA. PLoS ONE 2017, 12, e0185211. [Google Scholar] [CrossRef] [PubMed]
- Raymond, H.F.; Al-Tayyib, A.; Neaigus, A.; Reilly, K.H.; Braunstein, S.; Brady, K.A.; Sey, E.; Risser, J.; Padget, P.; Lalota, M.; et al. HIV Among MSM and Heterosexual Women in the United States: An Ecologic Analysis. J. Acquir. Immune Defic. Syndr. 2017, 75, S276–S280. [Google Scholar] [CrossRef] [PubMed]
- Saliou, A.; Delobel, P.; Dubois, M.; Nicot, F.; Calvez, V.; Masquelier, B.; Izopet, J.; the ANRS AC11 Resostamce Study Group. Concordance between Two Phenotypic Assays and Ultradeep Pyrosequencing for Determining HIV-1 Tropism. Antimicrob. Agents Chemother. 2011, 55, 2831–2836. [Google Scholar] [CrossRef]
- Prosperi, M.C.F.; Yin, L.; Nolan, D.J.; Lowe, A.D.; Goodenow, M.M.; Salemi, M. Empirical validation of viral quasispecies assembly algorithms: State-of-the-art and challenges. Sci. Rep. 2013, 3, 2837. [Google Scholar] [CrossRef]
- Mbondji-wonje, C.; Dong, M.; Wang, X.; Zhao, J.; Ragupathy, V.; Sanchez, A.M.; Denny, T.N.; Hewlett, I. Distinctive variation in the U3R region of the 5 ’ Long Terminal Repeat from diverse HIV-1 strains. PLoS ONE 2018, 13, e0195661. [Google Scholar] [CrossRef]
- Hiener, B.; Eden, J.; Horsburgh, B.A.; Palmer, S. Amplification of Near Full-length HIV-1 Proviruses for Next-Generation Sequencing. J. Vis. Exp. 2018, 140, e58016. [Google Scholar] [CrossRef]
- Hunt, M.; Gall, A.; Ong, S.H.; Brener, J.; Ferns, B.; Goulder, P.; Nastouli, E.; Keane, J.A.; Kellam, P.; Otto, T.D. IVA: Accurate de novo assembly of RNA virus genomes. Bioinformatics 2015, 31, 2374–2376. [Google Scholar] [CrossRef] [PubMed]
- Aralaguppe, S.G.; Siddik, A.B.; Manickam, A.; Ambikan, A.T.; Kumar, M.M.; Fernandes, S.J.; Amogne, W.; Bangaruswamy, D.K.; Hanna, L.E.; Sonnerborg, A.; et al. Multiplexed next-generation sequencing and de novo assembly to obtain near full-length HIV-1 genome from plasma virus. J. Virol Methods 2016, 236, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Alampalli, S.V.; Thomson, M.M.; Sampathkumar, R.; Sivaraman, K.; Anto Jesuraj, U.K.J.; Dhar, C.; Souza, G.D.; Berry, N.; Vyakarnam, A. Deep sequencing of near full-length HIV-1 genomes from plasma identifies circulating subtype C and infrequent occurrence of AC recombinant form in Southern India. PLoS ONE 2017, 12, e0188603. [Google Scholar] [CrossRef]
- Cornelissen, M.; Gall, A.; Vink, M.; Zorgdrager, F.; Edwards, S.; Jurriaans, S.; Bakker, M.; Ong, S.H.; Gras, L.; Van Sighem, A.; et al. From clinical sample to complete genome: Comparing methods for the extraction of HIV-1 RNA for high-throughput deep sequencing. Virus Res. 2017, 239, 10–16. [Google Scholar] [CrossRef]
- Ratmann, O.; Hodcroft, E.B.; Pickles, M.; Cori, A.; Hall, M.; Lycett, S.; Colijn, C.; Dearlove, B.; Didelot, X.; Frost, S.; et al. Phylogenetic Tools for Generalized HIV-1 Epidemics: Findings from the PANGEA-HIV Methods Comparison. Mol. Biol Evol. 2017, 34, 185–203. [Google Scholar] [CrossRef] [PubMed]
- Ratmann, O.; Grabowski, M.K.; Hall, M.; Golubchik, T.; Wymant, C.; Abeler-dörner, L.; Bonsall, D.; Hoppe, A.; Brown, A.L.; De Oliveira, T.; et al. Inferring HIV-1 transmission networks and sources of epidemic spread in Africa with deep-sequence phylogenetic analysis. Nat. Commun. 2019, 10, 1411. [Google Scholar] [CrossRef] [PubMed]
- Kafando, A.; Fournier, E.; Serhir, B.; Martineau, C.; Doualla-, F.; Sylla, M.; Chamberland, A.; El-, M.; Sangare, M.N.; Charest, H.; et al. HIV-1 envelope sequence-based diversity measures for identifying recent infections. PLoS ONE 2017, 12, e0189999. [Google Scholar] [CrossRef] [PubMed]
- Thomson, E.; Ip, C.L.C.; Badhan, A.; Christiansen, M.T.; Adamson, W.; Ansari, M.A.; Bibby, D.; Breuer, J.; Brown, A.; Bowden, R.; et al. Comparison of Next-Generation Sequencing Technologies for Comprehensive Assessment of Full-Length Hepatitis C Viral Genomes. J. Clin. Microbiol. 2016, 54, 2470–2484. [Google Scholar] [CrossRef] [PubMed]
- Welzel, T.M.; Bhardwaj, N.; Hedskog, C.; Chodavarapu, K.; Camus, G.; McNally, J.; Brainard, D.; Miller, M.D.; Mo, H.; Svarovskaia, E.; et al. Global epidemiology of HCV subtypes and resistance-associated substitutions evaluated by sequencing-based subtype analyses. J. Hepatol. 2017, 67, 224–236. [Google Scholar] [CrossRef]
- Ishida, Y.; Hayashida, T.; Sugiyama, M.; Tsuchiya, K.; Kikuchi, Y.; Mizokami, M.; Oka, S.; Gatanaga, H. Full-Genome Analysis of Hepatitis C Virus in Japanese and Non-Japanese Patients Coinfected with HIV-1 in Tokyo. J. Acquir. Immune Defic. Syndr. 2019, 80, 350–357. [Google Scholar] [CrossRef]
- Nguyen, T.; Delaugerre, C.; Valantin, M.-A.; Amiel, C.; Netzer, E.; Lʼyavanc, T.; Ohayon, M.; Valin, N.; Day, N.; Kreplak, G.; et al. Shared HCV Transmission Networks Among HIV-1–Positive and HIV-1–Negative Men Having Sex With Men by Ultradeep Sequencing. JAIDS J. Acquir. Immune Defic. Syndr. 2019, 82, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, S.R.; Kim, K.W.; Cheng, J.X.; Bull, R.A.; Stelzer-Braid, S.; Luciani, F.; Rawlinson, W.D.; Craig, M.E. Amplification and next generation sequencing of near full-length human enteroviruses for identification and characterisation from clinical samples. Sci. Rep. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Majumdar, M.; Martin, J. Detection by direct next generation sequencing analysis of emerging enterovirus D68 and C109 strains in an environmental sample from Scotland. Front. Microbiol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Gaspareto, K.V.; Ribeiro, R.M.; de Mello Malta, F.; Gomes-Gouvêa, M.S.; Muto, N.H.; Mendes-Correa, M.C.; Rozanski, A.; Carrilho, F.J.; Sabino, E.C.; Pinho, J.R.R. HCV inter-subtype 1a/1b recombinant detected by complete-genome next-generation sequencing. Arch. Virol. 2016, 161, 2161–2168. [Google Scholar] [CrossRef] [PubMed]
- Stelzl, E.; Haas, B.; Bauer, B.; Zhang, S.; Fiss, E.H.; Hillman, G.; Hamilton, A.T.; Mehta, R.; Heil, M.L.; Marins, E.G.; et al. First identification of a recombinant form of hepatitis C virus in Austrian patients by full-genome next generation sequencing. PLoS ONE 2017, 12, e0181273. [Google Scholar] [CrossRef]
- Ogishi, M.; Yotsuyanagi, H.; Tsutsumi, T.; Gatanaga, H.; Ode, H.; Sugiura, W.; Moriya, K.; Oka, S.; Kimura, S.; Koike, K. Deconvoluting the composition of low-frequency hepatitis C viral quasispecies: Comparison of genotypes and NS3 resistance-associated variants between HCV/HIV coinfected hemophiliacs and HCV monoinfected patients in Japan. PLoS ONE 2015, 10, e0119145. [Google Scholar] [CrossRef]
- Rodrigo, C.; Leung, P.; Lloyd, A.R.; Bull, R.A.; Luciani, F.; Grebely, J.; Dore, G.J.; Applegate, T.; Page, K.; Bruneau, J.; et al. Genomic variability of within-host hepatitis C variants in acute infection. J. Viral Hepat. 2019, 26, 476–484. [Google Scholar] [CrossRef]
- Abayasingam, A.; Leung, P.; Eltahla, A.; Bull, R.A.; Luciani, F.; Grebely, J.; Dore, G.J.; Applegate, T.; Page, K.; Bruneau, J.; et al. Genomic characterization of hepatitis C virus transmitted founder variants with deep sequencing. Infect. Genet. Evol. 2019, 71, 36–41. [Google Scholar] [CrossRef]
- Eltahla, A.A.; Leung, P.; Pirozyan, M.R.; Rodrigo, C.; Grebely, J.; Applegate, T.; Maher, L.; Luciani, F.; Lloyd, A.R.; Bull, R.A. Dynamic evolution of hepatitis C virus resistance-associated substitutions in the absence of antiviral treatment. Sci. Rep. 2017, 7, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Iles, J.C.; Njouom, R.; Foupouapouognigni, Y.; Bonsall, D.; Bowden, R.; Trebes, A.; Piazza, P.; Barnes, E.; Pépin, J.; Klenerman, P.; et al. Characterization of hepatitis c virus recombination in Cameroon by use of nonspecific next-generation sequencing. J. Clin. Microbiol. 2015, 53, 3155–3164. [Google Scholar] [CrossRef] [PubMed]
- Bendall, M.L.; Gibson, K.M.; Steiner, M.C.; Pérez-Losada, M.; Keith, A. Crandall HAPHPIPE: Haplotype reconstruction and real-time phylodynamics for deep sequencing of intra-host viral populations. Mol. Biol. Evol. 2020, (in press).
- Ambler, S.W. Agile Modeling; John Wiley & Sons: Hoboken, NJ, USA, 2002. [Google Scholar]
- Bertels, F.; Leemann, C.; Metzner, K.J.; Regoes, R.R. Parallel Evolution of HIV-1 in a Long-Term Experiment. Mol. Biol. Evol. 2019, 36, 2400–2414. [Google Scholar] [CrossRef] [PubMed]
- Babcock, G.J.; Iyer, S.; Smith, H.L.; Wang, Y.; Rowley, K.; Ambrosino, D.M.; Zamore, P.D.; Pierce, B.G.; Molrine, D.C.; Weng, Z. High-throughput sequencing analysis of post-liver transplantation HCV E2 glycoprotein evolution in the presence and absence of neutralizing monoclonal antibody. PLoS ONE 2014, 9, e0100325. [Google Scholar] [CrossRef] [PubMed]
- Eliseev, A.; Gibson, K.M.; Avdeyev, P.; Novik, D.; Bendall, M.L.; Pérez-Losada, M.; Alexeev, N.; Crandall, K.A. Evaluation of haplotype callers for next-generation sequencing of viruses. Infect. Genet. Evol. 2020, 82, 104277. [Google Scholar] [CrossRef] [PubMed]
- Noguera-Julian, M.; Edgil, D.; Harrigan, P.R.; Sandstrom, P.; Godfrey, C.; Paredes, R. Next-Generation Human Immunodeficiency Virus Sequencing for Patient Management and Drug Resistance Surveillance. J. Infect. Dis. 2017, 216, S829–S833. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Alnafee, K. Versatile and open software for comparing large genomes. Genome Biol. 2016, 5, 12–23. [Google Scholar] [CrossRef]
- Prabhakaran, S.; Rey, M.; Zagordi, O.; Beerenwinkel, N.; Roth, V. HIV haplotype inference using a propagating dirichlet process mixture model. IEEE/ACM Trans. Comput. Biol. Bioinform. 2014, 11, 182–191. [Google Scholar] [CrossRef]
- Knyazev, S.; Tsyvina, V.; Melnyk, A.; Malygina, T.; Porozov, Y.B.; Campbell, E.; Switzer, W.M.; Skums, P.; Zelikovsky, A. CliqueSNV: Scalable Reconstruction of Intra-Host Viral Populations from NGS Reads. bioRxiv 2018, 1–8. [Google Scholar] [CrossRef]
- Nikolenko, S.I.; Korobeynikov, A.I.; Alekseyev, M.A. BayesHammer: Bayesian clustering for error correction in single-cell sequencing. BMC Genom. 2013, 14. [Google Scholar] [CrossRef]
- Nurk, S.; Bankevich, A.; Antipov, D.; Gurevich, A.A.; Korobeynikov, A.; Lapidus, A.; Prjibelski, A.D.; Pyshkin, A.; Sirotkin, A.; Sirotkin, Y.; et al. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J. Comput. Biol. 2013, 20, 714–737. [Google Scholar] [CrossRef] [PubMed]
- Leal, E.; Villanova, F.E. Diversity of HIV-1 Subtype B: Implications to the Origin of BF Recombinants. PLoS ONE 2010, 5, e011833. [Google Scholar] [CrossRef] [PubMed]
- Ibe, S.; Shigemi, U.; Sawaki, K.; Fujisaki, S.; Hattori, J.; Yokomaku, Y.; Mamiya, N.; Hamaguchi, M.; Kaneda, T. Analysis of near full-length genomic sequences of drug-resistant HIV-1 spreading among therapy-naive individuals in Nagoya, Japan: Amino acid mutations associated with viral replication activity. AIDS Res. Hum. Retrovir. 2008, 24, 1121–1125. [Google Scholar] [CrossRef] [PubMed]
- Oelrichs, R.; Tsykin, A.; Rhodes, D.; Solomon, A.; Ellett, A.; McPhee, D.; Deacon, N. Genomic sequence of HIV type 1 from four members of the Sydney Blood Bank Cohort of long-term nonprogressors. AIDS Res. Hum. Retrovir. 1998, 14, 811–814. [Google Scholar] [CrossRef]
- Novelli, P.; Vella, C.; Oxford, J.; Daniels, R.S. Construction and biological characterization of an infectious molecular clone of HIV type 1GB8. AIDS Res. Hum. Retrovir. 2000, 16, 1175–1178. [Google Scholar] [CrossRef]
- Hierholzer, J.; Montano, S.; Hoelscher, M.; Negrete, M.; Hierholzer, M.; Avila, M.M.; Carrillo, M.G.; Russi, J.C.; Vinoles, J.; Alava, A.; et al. Molecular Epidemiology of HIV Type 1 in Ecuador, Peru, Bolivia, Uruguay, and Argentina. AIDS Res. Hum. Retrovir. 2002, 18, 1339–1350. [Google Scholar] [CrossRef]
- Viñoles, J.; Serra, M.; Russi, J.C.; Ruchansky, D.; Sosa-Estani, S.; Montano, S.M.; Carrion, G.; Eyzaguirre, L.M.; Carr, J.K.; Olson, J.G.; et al. Seroincidence and phylogeny of human immunodeficiency virus infections in a cohort of commercial sex workers in Montevideo, Uruguay. Am. J. Trop. Med. Hyg. 2005, 72, 495–500. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saad, M.D.; Al-Jaufy, A.; Grahan, R.R.; Nadai, Y.; Earhart, K.C.; Sanchez, J.L.; Carr, J.K. HIV type 1 strains common in Europe, Africa, and Asia cocirculate in Yemen. AIDS Res. Hum. Retrovir. 2005, 21, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Mikhail, M.; Wang, B.; Lemey, P.; Beckthold, B.; Vandamme, A.M.; Gill, M.J.; Saksena, N.K. Role of viral evolutionary rate in HIV-1 disease progression in a linked cohort. Retrovirology 2005, 2, 1–10. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Allen, T.M.; Altfeld, M.; Geer, S.C.; Kalife, E.T.; Moore, C.; Desouza, I.; Feeney, M.E.; Eldridge, R.L.; Maier, E.L.; Kaufmann, D.E.; et al. Selective Escape from CD8+ T-Cell Responses Represents a Major Driving Force of Human Immunodeficiency Virus Type 1 (HIV-1) Sequence Diversity and Reveals Constraints on HIV-1 Evolution. J. Virol. 2005, 79, 13239–13249. [Google Scholar] [CrossRef]
- Li, B.; Gladden, A.D.; Altfeld, M.; Kaldor, J.M.; Cooper, D.A.; Kelleher, A.D.; Allen, T.M. Rapid Reversion of Sequence Polymorphisms Dominates Early Human Immunodeficiency Virus Type 1 Evolution. J. Virol. 2007, 81, 193–201. [Google Scholar] [CrossRef]
- Frahm, N.; Kaufmann, D.E.; Yusim, K.; Muldoon, M.; Kesmir, C.; Linde, C.H.; Fischer, W.; Allen, T.M.; Li, B.; McMahon, B.H.; et al. Increased Sequence Diversity Coverage Improves Detection of HIV-Specific T Cell Responses. J. Immunol. 2007, 179, 6638–6650. [Google Scholar] [CrossRef]
- Andresen, B.S.; Vinner, L.; Tang, S.; Bragstad, K.; Kronborg, G.; Gerstoft, J.; Corbet, S.; Fomsgaard, A. Characterization of near full-length genomes of HIV type 1 strains in Denmark: Basis for a universal therapeutic vaccine. AIDS Res. Hum. Retrovir. 2007, 23, 1442–1448. [Google Scholar] [CrossRef]
- Nadai, Y.; Eyzaguirre, L.M.; Sill, A.; Cleghorn, F.; Nolte, C.; Charurat, M.; Collado-Chastel, S.; Jack, N.; Bartholomew, C.; Pape, J.W.; et al. HIV-1 epidemic in the Caribbean is dominated by subtype B. PLoS ONE 2009, 4, e04814. [Google Scholar] [CrossRef] [PubMed]
- Diaz, R.S.; Leal, É.; Sanabani, S.; Sucupira, M.C.A.; Tanuri, A.; Sabino, E.C.; Janini, L.M. Selective regimes and evolutionary rates of HIV-1 subtype B V3 variants in the Brazilian epidemic. Virology 2008, 381, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Kousiappa, I.; Van De Vijver, D.A.M.C.; Kostrikis, L.G. Near full-length genetic analysis of HIV sequences derived from Cyprus: Evidence of a highly polyphyletic and evolving infection. AIDS Res. Hum. Retrovir. 2009, 25, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.E.; Li, B.; Carlson, J.M.; Streeck, H.; Gladden, A.D.; Goodman, R.; Schneidewind, A.; Power, K.A.; Toth, I.; Frahm, N.; et al. Protective HLA Class I Alleles That Restrict Acute-Phase CD8+ T-Cell Responses Are Associated with Viral Escape Mutations Located in Highly Conserved Regions of Human Immunodeficiency Virus Type 1. J. Virol. 2009, 83, 1845–1855. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Gonzalez, J.F.; Salazar, M.G.; Keele, B.F.; Learn, G.H.; Giorgi, E.E.; Li, H.; Decker, J.M.; Wang, S.; Baalwa, J.; Kraus, M.H.; et al. Genetic identity, biological phenotype, and evolutionary pathways of transmitted/founder viruses in acute and early HIV-1 infection. J. Exp. Med. 2009, 206, 1273–1289. [Google Scholar] [CrossRef]
- Cuevas, M.T.; Fernandez-Garcia, A.; Pinilla, M.; Garcia-Alvarez, V.; Thomson, M.; Delgado, E.; Gonzalez-Galeano, M.; Miralles, C.; Serrano-Bengoechea, E.; Ojea de Castro, R.; et al. Short communication: Biological and genetic characterization of HIV type 1 subtype B and nonsubtype B transmitted viruses: Usefulness for vaccine candidate assessment. AIDS Res. Hum. Retrovir. 2010, 26, 1019–1025. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, E.L.; Wong, M.; Wang, S.; Wei, X.; Jones, N.A.; Conrod, K.E.; Aldam, D.; Turner, J.; Pellegrino, P.; Keele, B.F.; et al. Kinetics of Expansion of Epitope-Specific T Cell Responses during Primary HIV-1 Infection. J. Immunol. 2009, 182, 7131–7145. [Google Scholar] [CrossRef]
- Rolland, M.; Tovanabutra, S.; Allan, C.; Frahm, N.; Peter, B.; Sanders-buell, E.; Heath, L.; Magaret, C.A.; Bose, M.; Sullivan, A.O.; et al. Genetic impact of vaccination on breakthrough HIV-1 sequences from the Step trial. Nat. Med. 2011, 17, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Kousiappa, I.; Achilleos, C.; Hezka, J.; Lazarou, Y.; Othonos, K.; Demetriades, I.; Kostrikis, L.G. Molecular characterization of HIV type 1 strains from newly diagnosed patients in Cyprus (2007–2009) recovers multiple clades including unique recombinant strains and lack of transmitted drug resistance. AIDS Res. Hum. Retrovir. 2011, 27, 1183–1199. [Google Scholar] [CrossRef]
- Eyzaguirrea, L.M.; Charurata, M.; Redfielda, R.R.; Blattnera, W.A.; Carra, J.K.; Sajadi, M.M. Elevated hypermutation levels in HIV-1 natural viral suppressors. Virology 2013, 443, 306–312. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, Z.; He, X.; Wang, Z.; Xing, H.; Li, F.; Yang, Y.; Wang, Q.; Takebe, Y.; Shao, Y. Tracing the origin and history of HIV-1 subtype B′ epidemic by near full-length genome analyses. Aids 2012, 26, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Kijak, G.H.; Tovanabutra, S.; Rerks-Ngarm, S.; Nitayaphan, S.; Eamsila, C.; Kunasol, P.; Khamboonruang, C.; Thongcharoen, P.; Namwat, C.; Premsri, N.; et al. Molecular Evolution of the HIV-1 Thai Epidemic between the Time of RV144 Immunogen Selection to the Execution of the Vaccine Efficacy Trial. J. Virol. 2013, 87, 7265–7281. [Google Scholar] [CrossRef] [PubMed]
- Sanabani, S.S.; de Pastena, É.R.S.; da Costa, A.C.; Martinez, V.P.; Kleine-Neto, W.; de Oliveira, A.C.S.; Sauer, M.M.; Bassichetto, K.C.; Oliveira, S.M.S.; Tomiyama, H.T.I.; et al. Characterization of partial and near full-length genomes of HIV-1 strains sampled from recently infected individuals in São Paulo, Brazil. PLoS ONE 2011, 6, e025869. [Google Scholar] [CrossRef] [PubMed]
- Henn, M.R.; Boutwell, C.L.; Charlebois, P.; Lennon, N.J.; Power, K.A.; Macalalad, A.R.; Berlin, A.M.; Malboeuf, C.M.; Ryan, E.M.; Gnerre, S.; et al. Whole genome deep sequencing of HIV-1 reveals the impact of early minor variants upon immune recognition during acute infection. PLoS Pathog. 2012, 8, e1002529. [Google Scholar] [CrossRef]
- Edlefsen, P.T.; Rolland, M.; Hertz, T.; Tovanabutra, S.; Gartland, A.J.; deCamp, A.C.; Magaret, C.A.; Ahmed, H.; Gottardo, R.; Juraska, M.; et al. Comprehensive Sieve Analysis of Breakthrough HIV-1 Sequences in the RV144 Vaccine Efficacy Trial. PLoS Comput. Biol. 2015, 11, 1–37. [Google Scholar] [CrossRef]
- An, M.; Han, X.; Xu, J.; Chu, Z.; Jia, M.; Wu, H.; Lu, L.; Takebe, Y.; Shang, H. Reconstituting the Epidemic History of HIV Strain CRF01_AE among Men Who Have Sex with Men (MSM) in Liaoning, Northeastern China: Implications for the Expanding Epidemic among MSM in China. J. Virol. 2012, 86, 12402–12406. [Google Scholar] [CrossRef]
- Sanchez-Pescador, R.; Power, M.D.; Barr, P.J.; Steimer, K.S.; Stempien, M.M.; Brown-Shimer, S.L.; Gee, W.W.; Renard, A.; Randolph, A.; Levy, J.A. Nucleotide sequence and expression of an AIDS-associated retrovirus (ARV-2). Science 1985, 227, 484–492. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; An, M.; Zhao, B.; Duan, S.; Yang, S.; Xu, J.; Zhang, M.; McGoogan, J.M.; Takebe, Y.; Shang, H. High Prevalence of HIV-1 Intersubtype B′/C Recombinants among Injecting Drug Users in Dehong, China. PLoS ONE 2013, 8, e065337. [Google Scholar] [CrossRef] [PubMed]
- Salgado, M.; Swanson, M.D.; Pohlmeyer, C.W.; Buckheit, R.W.; Wu, J.; Archin, N.M.; Williams, T.M.; Margolis, D.M.; Siliciano, R.F.; Garcia, J.V.; et al. HLA-B*57 Elite Suppressor and Chronic Progressor HIV-1 Isolates Replicate Vigorously and Cause CD4+ T Cell Depletion in Humanized BLT Mice. J. Virol. 2014, 88, 3340–3352. [Google Scholar] [CrossRef]
- Cho, Y.K.; Kim, J.E.; Foley, B.T. Phylogenetic analysis of near full-length HIV type 1 genomic sequences from 21 korean individuals. AIDS Res. Hum. Retrovir. 2013, 29, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Pessôa, R.; Watanabe, J.T.; Calabria, P.; Alencar, C.S.; Loureiro, P.; Lopes, M.E.; Proetti, A.B.; Félix, A.C.; Ester, C. International Component of the NHLBI Epidemiology and Donor Evaluation Study-III (REDS-III) Enhanced detection of viral diversity using partial and near full- length genomes of HIV-1 provirus deep sequencing data from recently infected donors at four blood centers in Brazil. Transfusion 2015, 55, 980–990. [Google Scholar] [CrossRef] [PubMed]
- Grossmann, S.; Nowak, P.; Neogi, U. Subtype-independent near full-length HIV-1 genome sequencing and assembly to be used in large molecular epidemiological studies and clinical management. J. Int. AIDS Soc. 2015, 18, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Blanco, M.; Machado, L.Y.; Díaz, H.; Ruiz, N.; Romay, D.; Silva, E. HIV-1 genetic variability in Cuba and implications for transmission and clinical progression. MEDICC Rev. 2015, 17, 25–31. [Google Scholar]
- Tully, D.C.; Ogilvie, C.B.; Batorsky, R.E.; Bean, D.J.; Power, K.A.; Ghebremichael, M.; Bedard, H.E.; Gladden, A.D.; Seese, A.M.; Amero, M.A.; et al. Differences in the Selection Bottleneck between Modes of Sexual Transmission Influence the Genetic Composition of the HIV-1 Founder Virus. PLoS Pathog. 2016, 12, 1–29. [Google Scholar] [CrossRef]
- Pessôa, R.; Loureiro, P.; Lopes, M.E.; Carneiro-Proietti, A.B.F.; Sabino, E.C.; Busch, M.P.; Sanabani, S.S. Ultra-deep sequencing of HIV-1 near full-length and partial proviral genomes reveals high genetic diversity among Brazilian blood donors. PLoS ONE 2016, 11, e0152499. [Google Scholar] [CrossRef]
- Bruner, K.M.; Murray, A.J.; Pollack, R.A.; Soliman, M.G.; Sarah, B.; Capoferri, A.A.; Lai, J.; Strain, M.C.; Lada, S.M.; Ho, Y.; et al. Defective proviruses rapidly accumulate during acute HIV-1 infection Katherine. Nat. Med. 2016, 22, 1043–1049. [Google Scholar] [CrossRef]
- Cevallos, C.G.; Jones, L.R.; Pando, M.A.; Carr, J.K.; Avila, M.M.; Quarleri, J. Genomic characterization and molecular evolution analysis of subtype B and BF recombinant HIV-1 strains among Argentinean men who have sex with men reveal a complex scenario. PLoS ONE 2017, 12, e0189705. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.K.; Fultz, P.N.; Keddie, E.; Saag, M.S.; Sharp, P.M.; Hahn, B.H.; Shaw, G.M. A Molecular Clone of HIV-1 Tropic and Cytopathic for Human and Chimpanzee Lymphocytes. Virology 1993, 194, 858–864. [Google Scholar] [CrossRef]
- Neogi, U.; Siddik, A.B.; Kalaghatgi, P.; Gisslén, M.; Bratt, G.; Marrone, G.; Sönnerborg, A. Recent increased identification and transmission of HIV-1 unique recombinant forms in Sweden. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Kreutz, R.; Dietrich, U.; Kuhnel, H.; Nieselt-Struwe, K.; Eigen, M.; Rubsamen-Waigmann, H. Analysis of the envelope region of the highly divergent HIV-2ALT isolate extends the known range of variability within the primate immunodeficiency viruses. AIDS Res. Hum. Retrovir. 1992, 8, 1619–1629. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.K.; Laukkanen, T.; Salminen, M.O.; Albert, J.; Alaeus, A.; Kim, B.; Sanders-Buell, E.; Birx, D.L.; McCutchan, F.E. Characterization of subtype A HIV-1 from Africa by full genome sequencing. Aids 1999, 13, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Laukkanen, T.; Albert, J.; Liitsola, K.; Green, S.D.; Carr, J.K.; Leitner, T.; McCutchan, F.E.; Salminen, M.O. Virtually full-length sequences of HIV type 1 subtype J reference strains. AIDS Res. Hum. Retrovir. 1999, 15, 293–297. [Google Scholar] [CrossRef] [PubMed]
- Hoelscher, M.; Kim, B.; Maboko, L.; Mhalu, F.; Von Sonnenburg, F.; Birx, D.L.; McCutchan, F.E. High proportion of unrelated HIV-1 intersubtype recombinants in the Mbeya region of southwest Tanzania. Aids 2001, 15, 1461–1470. [Google Scholar] [CrossRef]
- Novitsky, V.; Smith, U.R.; Gilbert, P.; McLane, M.F.; Chigwedere, P.; Williamson, C.; Ndung’u, T.; Klein, I.; Chang, S.Y.; Peter, T.; et al. Human Immunodeficiency Virus Type 1 Subtype C Molecular Phylogeny: Consensus Sequence for an AIDS Vaccine Design? J. Virol. 2002, 76, 5435–5451. [Google Scholar] [CrossRef]
- Harris, M.E.; Serwadda, D.; Sewankambo, N.; Kim, B.; Kigozi, G.; Kiwanuka, N.; Phillips, J.B.; Wabwire, F.; Meehen, M.; Lutalo, T.; et al. Among 46 near full length HIV type 1 genome sequences from Rakai District, Uganda, subtype D and AD recombinants predominate. AIDS Res. Hum. Retrovir. 2002, 18, 1281–1290. [Google Scholar] [CrossRef]
- Triques, K.; Bourgeois, A.; Vidal, N.; Mpoudi-Ngole, E.; Mulanga-Kabeya, C.; Nzilambi, N.; Torimiro, N.; Saman, E.; Delaporte, E.; Peeters, M. Near-full-length genome sequencing of divergent African HIV type 1 subtype F viruses leads to the identification of a new HIV type 1 subtype designated K. AIDS Res. Hum. Retrovir. 2000, 16, 139–151. [Google Scholar] [CrossRef]
- Arroyo, M.A.; Hoelscher, M.; Sanders-Buell, E.; Herbinger, K.-H.; Samky, E.; Maboko, L.; Hoffmann, O.; Robb, M.R.; Birx, D.L.; McCutchan, F.E. HIV type 1 subtypes among blood donors in the Mbeya region of southwest Tanzania. AIDS Res. Hum. Retrovir. 2004, 20, 895–901. [Google Scholar] [CrossRef] [PubMed]
- Kijak, G.H.; Sanders-Buell, E.; Wolfe, N.D.; Mpoudi-Ngole, E.; Kim, B.; Brown, B.; Robb, M.L.; Birx, D.L.; Burke, D.S.; Carr, J.K.; et al. Development and application of a high-throughput HIV type 1 genotyping assay to identify CRF02_AG in West/West Central Africa. AIDS Res. Hum. Retrovir. 2004, 20, 521–530. [Google Scholar] [CrossRef] [PubMed]
- Carrion, G.; Eyzaguirre, L.; Montano, S.M.; Laguna-Torres, V.; Serra, M.; Aguayo, N.; Avila, M.M.; Ruchansky, D.; Pando, M.A.; Vinoles, J.; et al. Documentation of subtype C HIV Type 1 strains in Argentina, Paraguay, and Uruguay. AIDS Res. Hum. Retrovir. 2004, 20, 1022–1025. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Z.; Xing, H.; Wei, M.; Duan, Y.; Zhao, Q.; Xu, J.; Shao, Y. Characterization of five nearly full-length genomes of early HIV type 1 strains in Ruili city: Implications for the genesis of CRF07_BC and CRF08_BC circulating in China. AIDS Res. Hum. Retrovir. 2005, 21, 1051–1056. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, C.M.; Birditt, B.A.; McKay, A.R.; Stoddard, J.N.; Lee, T.C.; McLaughlin, S.; Moore, S.W.; Shindo, N.; Learn, G.H.; Korber, B.T.; et al. Large-scale amplification, cloning and sequencing of near full-length HIV-1 subtype C genomes. J. Virol. Methods 2006, 136, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Abecasis, A.B.; Lemey, P.; Vidal, N.; de Oliveira, T.; Peeters, M.; Camacho, R.; Shapiro, B.; Rambaut, A.; Vandamme, A.-M. Recombination Confounds the Early Evolutionary History of Human Immunodeficiency Virus Type 1: Subtype G Is a Circulating Recombinant Form. J. Virol. 2007, 81, 8543–8551. [Google Scholar] [CrossRef] [PubMed]
- Tovanabutra, S.; Sanders, E.J.; Graham, S.M.; Mwangome, M.; Peshu, N.; McClelland, R.S.; Muhaari, A.; Crossler, J.; Price, M.A.; Gilmour, J.; et al. Evaluation of HIV type 1 strains in men having sex with men and in female sex workers in Mombasa, Kenya. AIDS Res. Hum. Retrovir. 2010, 26, 123–131. [Google Scholar] [CrossRef]
- Treurnicht, F.K.; Seoighe, C.; Martin, D.P.; Wood, N.; Abrahams, M.-R.; Rosa, D.d.A.; Bredell, H.; Woodman, Z.; Hide, W.; Mlisana, K.; et al. Adaptive changes in HIV-1 subtype C proteins during early infection are driven by changes in HLA-associated immune pressure. Virology 2010, 396, 213–225. [Google Scholar] [CrossRef][Green Version]
- Wilkinson, E.; Holzmayer, V.; Jacobs, G.B.; De Oliveira, T.; Brennan, C.A.; Hackett, J.; Van Rensburg, E.J.; Engelbrecht, S. Sequencing and phylogenetic analysis of near full-length HIV-1 subtypes A, B, G and unique recombinant AC and AD viral strains identified in South Africa. AIDS Res. Hum. Retrovir. 2015, 31, 412–420. [Google Scholar] [CrossRef]
- Tongo, M.; Dorfman, J.R.; Abrahams, M.R.; Mpoudi-Ngole, E.; Burgers, W.A.; Martin, D.P. Near full-length HIV type 1M genomic sequences from Cameroon. Evol. Med. Public Heal. 2015, 2015, 254–265. [Google Scholar] [CrossRef]
- Billings, E.; Sanders-Buell, E.; Bose, M.; Bradfield, A.; Lei, E.; Kijak, G.H.; Arroyo, M.A.; Kibaya, R.M.; Scott, P.T.; Wasunna, M.K.; et al. The number and complexity of pure and recombinant HIV-1 strains observed within incident infections during the HIV and malaria cohort study conducted in Kericho, Kenya, from 2003 to 2006. PLoS ONE 2015, 10, e0135124. [Google Scholar] [CrossRef] [PubMed]
- Amogne, W.; Bontell, I.; Grossmann, S.; Aderaye, G.; Lindquist, L.; Sonnerborg, A.; Neogi, U. Phylogenetic Analysis of Ethiopian HIV-1 Subtype C Near Full-Length Genomes Reveals High Intrasubtype Diversity and a Strong Geographical Cluster. AIDS Res. Hum. Retrovir. 2016, 32, 471–474. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Hora, B.; DeMarco, T.; Shah, S.A.; Ahmed, M.; Sanchez, A.M.; Su, C.; Carter, M.; Stone, M.; Hasan, R.; et al. Fast dissemination of new HIV-1 CRF02/A1 recombinants in Pakistan. PLoS ONE 2016, 11, e0167839. [Google Scholar] [CrossRef]
- Billings, E.; Sanders-Buell, E.; Bose, M.; Kijak, G.H.; Bradfield, A.; Crossler, J.; Arroyo, M.A.; Maboko, L.; Hoffmann, O.; Geis, S.; et al. HIV-1 Genetic Diversity Among Incident Infections in Mbeya, Tanzania. AIDS Res. Hum. Retrovir. 2017, 33, 373–381. [Google Scholar] [CrossRef]
- Rodgers, M.A.; Wilkinson, E.; Vallari, A.; McArthur, C.; Sthreshley, L.; Brennan, C.A.; Cloherty, G.; de Oliveira, T. Sensitive Next-Generation Sequencing Method Reveals Deep Genetic Diversity of HIV-1 in the Democratic Republic of the Congo. J. Virol. 2017, 91, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Li, L.; Myers, J.R.; Marth, G.T. ART: A next-generation sequencing read simulator. Bioinformatics 2012, 28, 593–594. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Strimmer, K.; von Haeseler, A.; Salemi, M. Genetic distances and nucleotide substitution models. Phylogenetic Handb. 2012, 111–141. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; RC Team: Vienna, Austria, 2013. [Google Scholar]
- RStudio Team. RStudio: Integrated Development for R.; RStudio Team: Boston, MA, USA, 2015. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- McDonald, J. Handbook of Biological Statistics, 3rd ed.; Sparky House Publishing: Baltimore, Maryland, 2014. [Google Scholar]
- Kruskal, W.H.; Wallis, A.W. Use of Ranks in One-Criterion Variance Analysis. J. Am. Stat. Assoc. 1952, 47, 583–621. [Google Scholar] [CrossRef]
- Dinno, A. Dunn.test: Dunn’s Test of Multiple Comparisons Using Rank Sums. R Package Version 2017, 1. [Google Scholar]
- Holm, S. A Simple Sequentially Rejective Multiple Test Procedure. Scand J. Stat. 1979, 6, 65–70. [Google Scholar]
- Woolson, R.F. Wilcoxon signed-rank test. In Wiley Encyclopedia of Clincal Trials; D’Agostino, R.B., Sullivan, L., Massaro, J., Eds.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 7–9. [Google Scholar]
- Pineda-Peña, A.C.; Faria, N.R.; Imbrechts, S.; Libin, P.; Abecasis, A.B.; Deforche, K.; Gómez-López, A.; Camacho, R.J.; De Oliveira, T.; Vandamme, A.M. Automated subtyping of HIV-1 genetic sequences for clinical and surveillance purposes: Performance evaluation of the new REGA version 3 and seven other tools. Infect. Genet. Evol. 2013, 19, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Kuiken, C.; Combet, C.; Bukh, J.; Shin-I, T.; Deleage, G.; Mizokami, M.; Richardson, R.; Sablon, E.; Yusim, K.; Pawlotsky, J.M.; et al. A comprehensive system for consistent numbering of HCV sequences, proteins and epitopes. Hepatology 2006, 44, 1355–1361. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef]
- Crandall, K.A.; Posada, D.; Vasco, D. Effective population sizes: Missing measures and missing concepts. Anim. Conserv. 1999, 2, 317–319. [Google Scholar] [CrossRef]
- Nickle, D.C.; Jensen, M.A.; Shriner, G.S.G.D.; Learn, G.H.; Rodrigo, A.G.; Mullins, J.I. Consensus and Ancestral State HIV Vaccines. Science 2003, 299, 1515–1519. [Google Scholar] [CrossRef] [PubMed]
- Gibson, K.; Jair, K.; Castel, A.D.; Bendall, M.L.; Wilbourn, B.; Jordan, J.A.; Crandall, K.A.; Pérez-Losada, M.; the DC Cohort Executive Committee. A cross-sectional study to characterize local HIV-1 dynamics in Washington, DC using next-generation sequencing. Sci. Rep. 2020, 10, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Shafer, R. Web Resources for HIV type 1 Genotypic-Resistance Test Interpretation. Clin. Infect. Dis 2006, 42, 1608–1618. [Google Scholar] [CrossRef]
- Rhee, S.Y. Human immunodeficiency virus reverse transcriptase and protease sequence database. Nucleic Acids Res. 2003, 31, 298–303. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Salmela, L.; Sahlin, K.; Mäkinen, V.; Tomescu, A.I. Gap filling as exact path length problem. J. Comput. Biol. 2016, 23, 347–361. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Charlebois, P.; Gnerre, S.; Coole, M.G.; Lennon, N.J.; Levin, J.Z.; Qu, J.; Ryan, E.M.; Zody, M.C.; Henn, M.R. De novo assembly of highly diverse viral populations. BMC Genom. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]





| Pipeline | Haphpipe_Assemble_01 | Haphpipe_Assemble_02 | HyRDA | Geneious | Geneious |
|---|---|---|---|---|---|
| Assembly approach | De novo | Reference | Reference | Reference | De novo |
| Reference | None | Flexible | HXB2 | Flexible | None |
| Consensus refinement | Available | Available | No | Available | No |
| Genes | All | All | pol | All | All |
| Use | Free, Command line | Free, Command line | Free, Web-based | Paid GUI | Paid GUI |
| Read trimming | Available, Trimmomatic | Available, Trimmomatic | Yes, parameters set by user | Available, modified-Mott algorithm | Available, modified-Mott algorithm |
| Error correction | Available, SPAdes | Available, SPAdes | Set sequencing platform error rate | Available, BBNorm * | Available, BBNorm * |
| Tool | N | Avg Bowtie2 Alignment First Step | Avg Bowtie2 Alignment Last Step | Number of Samples Refine.02 | Number of Samples Refine.03 | Number of Samples Refine.04 | Number of Samples Refine.05 |
|---|---|---|---|---|---|---|---|
| Simulated HIV data subtype B | |||||||
| HP01 | 100 | 99.87% | 99.64% | 3 | 0 | 0 | 0 |
| HP02 | 100 | 88.95% | 95.67% | 100 | 93 | 2 | 0 |
| GDN | 100 | NA | 64.20% | NA | NA | NA | NA |
| GRB | 100 | NA | 82.63% | NA | NA | NA | NA |
| Simulated HIV data non-subtype B | |||||||
| HP01 | 50 | 98.80% | 96.92% | 50 | 0 | 0 | 0 |
| HP02 | 50 | 59.37% | 80.47% | 50 | 44 | 2 | 0 |
| GDN | 50 | NA | 64.30% | NA | NA | NA | NA |
| GRB | 50 | NA | 63.05% | NA | NA | NA | NA |
| Tool | Num of Seqs | Avg Bowtie2 Alignment First Step | Avg Bowtie2 Alignment Last Step | Number of Samples Refine.02 | Number of Samples Refine.03 | Number of Samples Refine.04 | Number of Samples Refine.05 |
|---|---|---|---|---|---|---|---|
| Empirical HIV Data | |||||||
| HP01 | 36 | 53.67% | 44.23% | 36 | 10 | 0 | 0 |
| HP02 | 36 | 54.69% | 47.58% | 36 | 5 | 0 | 0 |
| GDN | 36 | NA | 89.98% | NA | NA | NA | NA |
| GRB | 36 | NA | 27.68% | NA | NA | NA | NA |
| Empirical HCV Data | |||||||
| HP01 | 23 | 90.46% | 72.40% | 23 | 19 | 7 | 1 |
| HP02 | 23 | 78.13% | 75.27% | 23 | 22 | 4 | 1 |
| GDN *^ | 23 | NA | 97.27% | NA | NA | NA | NA |
| GRB | 23 | NA | 63.31% | NA | NA | NA | NA |
| Empirical SARS-CoV-2 Data | |||||||
| HP01 | 4 | 100% | 94.32% | 4 | 3 | 0 | 0 |
| HP02 | 4 | 100% | 94.36% | 4 | 4 | 0 | 0 |
| GDN *^ | 4 | NA | 80.55% | NA | NA | NA | NA |
| GRB * | 4 | NA | 94.23% | NA | NA | NA | NA |
| PRRT | int | gp120 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pipeline | N | H | S | π | θ | N | H | S | π | θ | N | H | S | π | θ |
| HP01 | 36 | 7 | 6 | 0.0005 | 0.0008 | 36 | 5 | 5 | 0.0009 | 0.0013 | 36 | 9 | 10 | 0.0012 | 0.0015 |
| HP02 | 36 | 6 | 6 | 0.0005 | 0.0009 | 36 | 5 | 5 | 0.0009 | 0.0014 | 36 | 10 | 13 | 0.0016 | 0.0016 |
| HP01 haplotypes | 132 | 78 | 119 | 0.0028 | 0.0127 | 162 | 52 | 35 | 0.0021 | 0.0064 | 224 | 167 | 149 | 0.0049 | 0.0154 |
| HP02 haplotypes | 139 | 85 | 110 | 0.0034 | 0.0123 | 140 | 46 | 34 | 0.0021 | 0.0071 | 70 | 40 | 61 | 0.0026 | 0.0067 |
| HyDRA | 36 | 1 | 2 | 0.0003 | 0.0003 | 36 | 1 | 2 | 0.0003 | 0.0006 | NA | NA | NA | NA | NA |
| GRB | 36 | 1 | 3 | 0.0004 | 0.0005 | 36 | 1 | 3 | 0.0004 | 0.0009 | 36 | 1 | 5 | 0.0006 | 0.0007 |
| GDN | 36 | 2 | 4 | 0.0005 | 0.0006 | 36 | 2 | 4 | 0.0007 | 0.0011 | 36 | 3 | 9 | 0.0009 | 0.0011 |
| core | E1 | E2 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pipeline | N | H | S | π | θ | N | H | S | π | θ | N | H | S | π | θ |
| HP01 | 23 | 16 | 33 | 0.0360 | 0.0379 | 23 | 21 | 188 | 0.0814 | 0.0793 | 23 | 20 | 406 | 0.1047 | 0.0960 |
| HP02 | 23 | 16 | 26 | 0.0373 | 0.0366 | 23 | 20 | 151 | 0.0703 | 0.0707 | 23 | 23 | 380 | 0.1028 | 0.0960 |
| HP01 haplotypes | 63 | 32 | 46 | 0.0328 | 0.0407 | 48 | 36 | 216 | 0.0792 | 0.0739 | 61 | 53 | 462 | 0.1021 | 0.0845 |
| HP02 haplotypes | 67 | 30 | 114 | 0.0619 | 0.1241 | 49 | 32 | 228 | 0.0812 | 0.0842 | 56 | 44 | 615 | 0.1213 | 0.1209 |
| GRB | 23 | 2 | 377 | 0.4561 | 0.3493 | 23 | 5 | 167 | 0.0736 | 0.0939 | 23 | 3 | 482 | 0.1236 | 0.1484 |
| GDN | 23 | 14 | 55 | 0.0826 | 0.0746 | 23 | 16 | 146 | 0.0678 | 0.0691 | 23 | 22 | 389 | 0.1051 | 0.0991 |
| Diversity Estimates | Adjusted Genetic p-Distance | |||||||
|---|---|---|---|---|---|---|---|---|
| Pipeline | N | H | S | π | θ | Average | STDEV | |
| HP01 | 4 | 1 | 0 | 0 | 0 | HP01 Initial | 0.0033 | 0.0035 |
| HP02 | 4 | 1 | 0 | 0 | 0 | HP01 Final | 0.0035 | 0.0034 |
| GDN | 4 | 4 | 371 | 0.0081 | 0.0083 | HP02 Initial | 0.0019 | 0.0015 |
| GRB | 4 | 1 | 13 | 0.0004 | 0.0004 | HP02 Final | 0.0021 | 0.0019 |
| GDN | 0.0417 | 0.0423 | ||||||
| GRB | 0.0031 | 0.0008 | ||||||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gibson, K.M.; Steiner, M.C.; Rentia, U.; Bendall, M.L.; Pérez-Losada, M.; Crandall, K.A. Validation of Variant Assembly Using HAPHPIPE with Next-Generation Sequence Data from Viruses. Viruses 2020, 12, 758. https://doi.org/10.3390/v12070758
Gibson KM, Steiner MC, Rentia U, Bendall ML, Pérez-Losada M, Crandall KA. Validation of Variant Assembly Using HAPHPIPE with Next-Generation Sequence Data from Viruses. Viruses. 2020; 12(7):758. https://doi.org/10.3390/v12070758
Chicago/Turabian StyleGibson, Keylie M., Margaret C. Steiner, Uzma Rentia, Matthew L. Bendall, Marcos Pérez-Losada, and Keith A. Crandall. 2020. "Validation of Variant Assembly Using HAPHPIPE with Next-Generation Sequence Data from Viruses" Viruses 12, no. 7: 758. https://doi.org/10.3390/v12070758
APA StyleGibson, K. M., Steiner, M. C., Rentia, U., Bendall, M. L., Pérez-Losada, M., & Crandall, K. A. (2020). Validation of Variant Assembly Using HAPHPIPE with Next-Generation Sequence Data from Viruses. Viruses, 12(7), 758. https://doi.org/10.3390/v12070758