The Dynamic Life of Virus Capsids


Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Rhinoviruses
2.1. The Canyon and Capsid “Breathing”
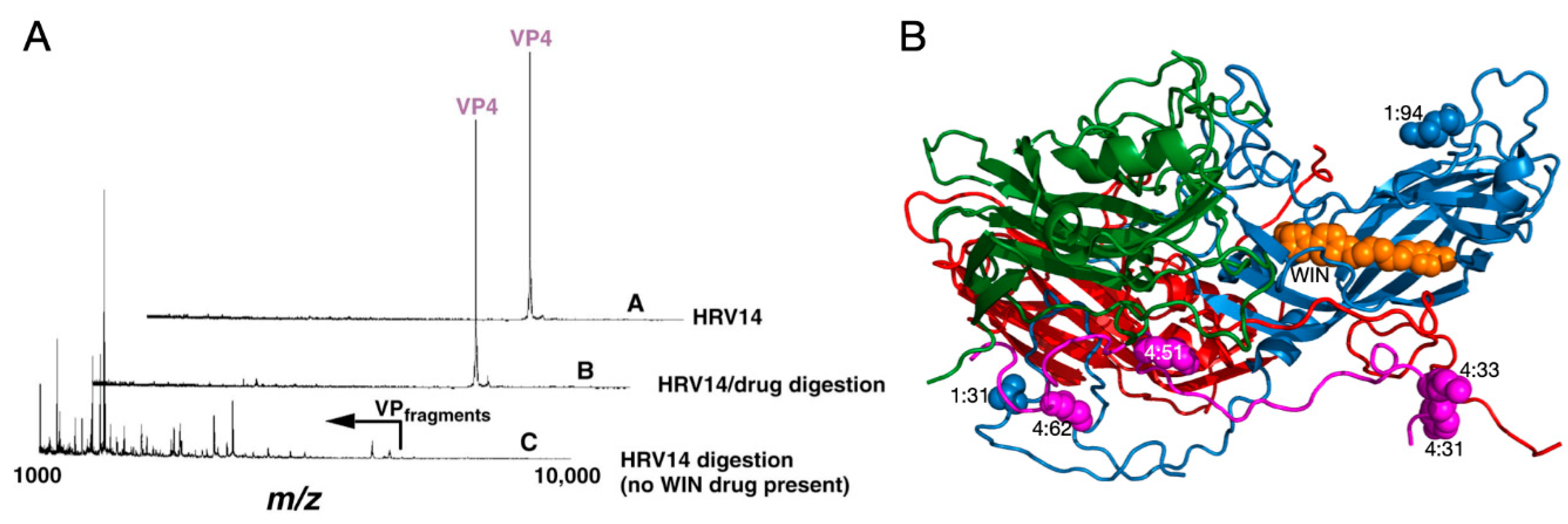
2.1.1. Mechanism of Antiviral Compounds
2.1.2. Antiviral Binding Cavity and “Pocket Factors”
2.1.3. Antiviral Resistant Mutants
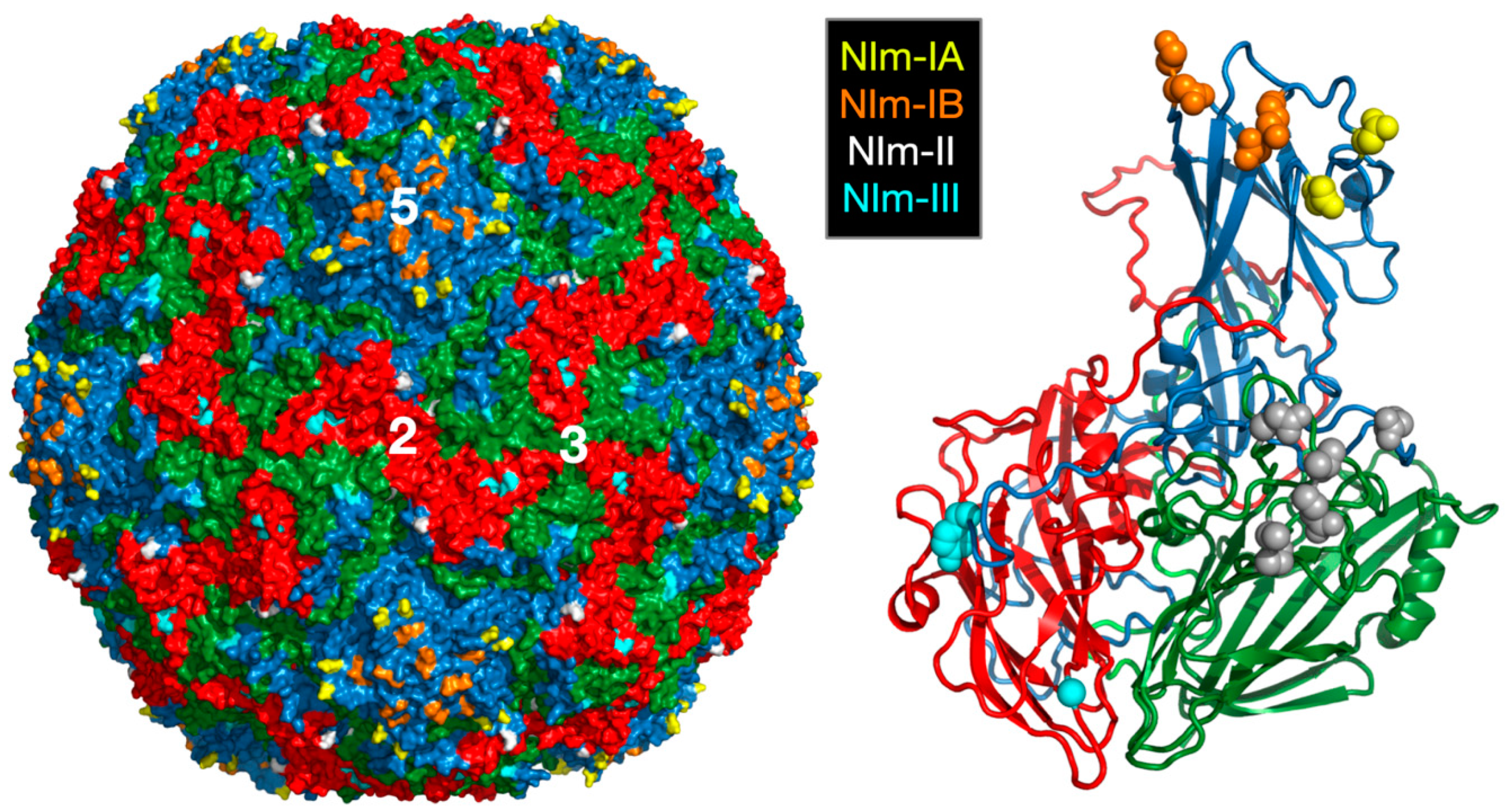
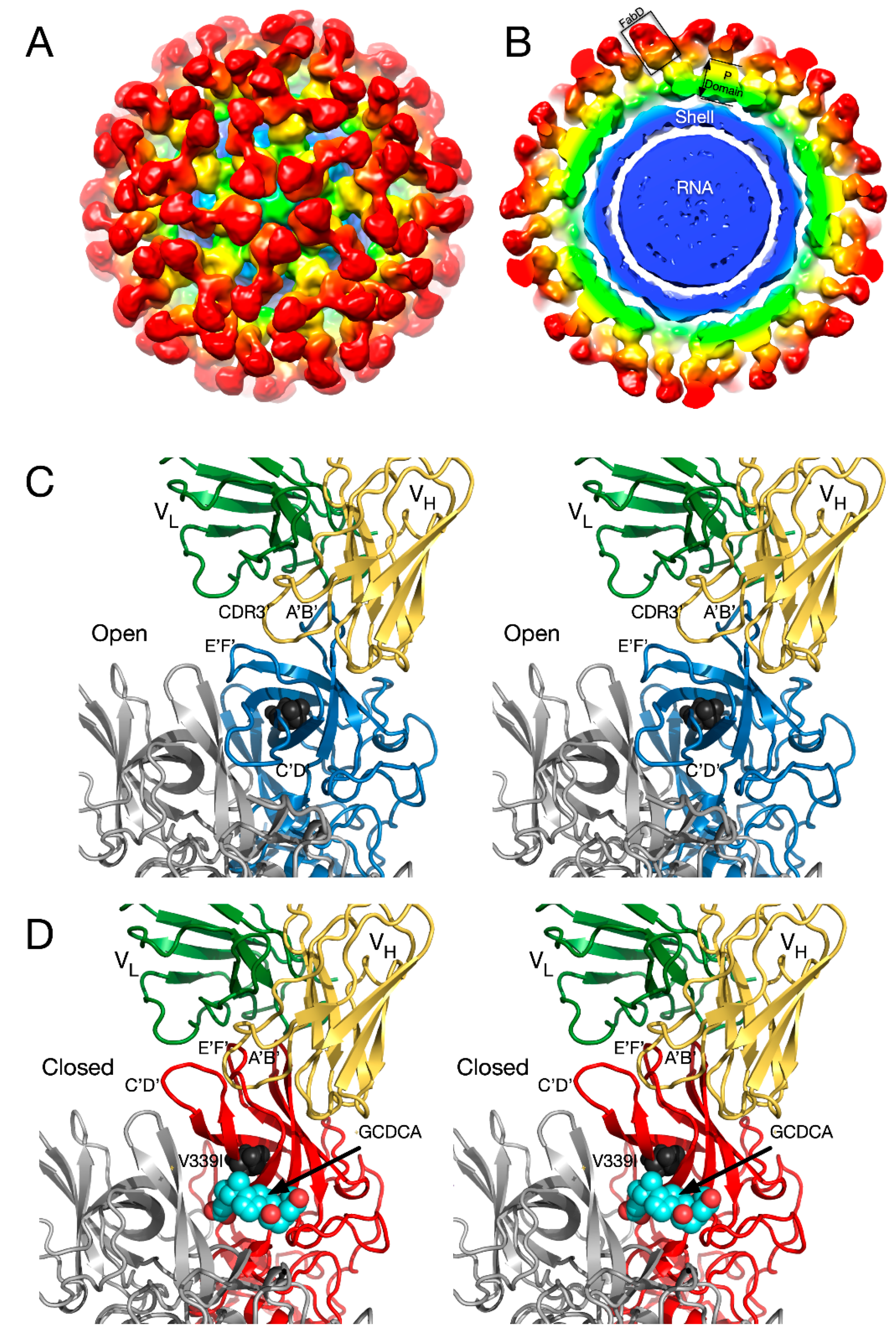
2.2. Antibody Neutralization of HRV and the Role of Capsid Flexibility
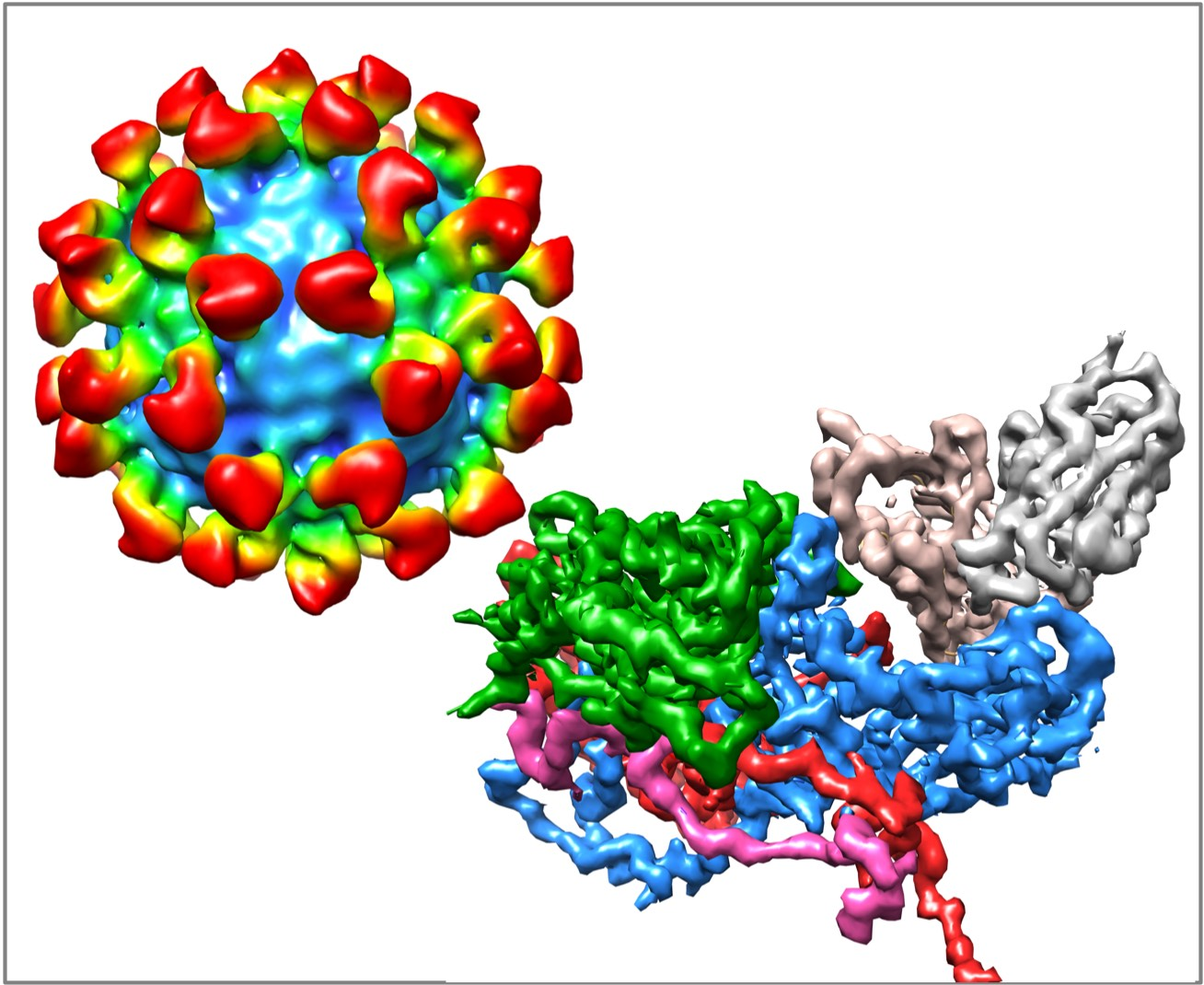
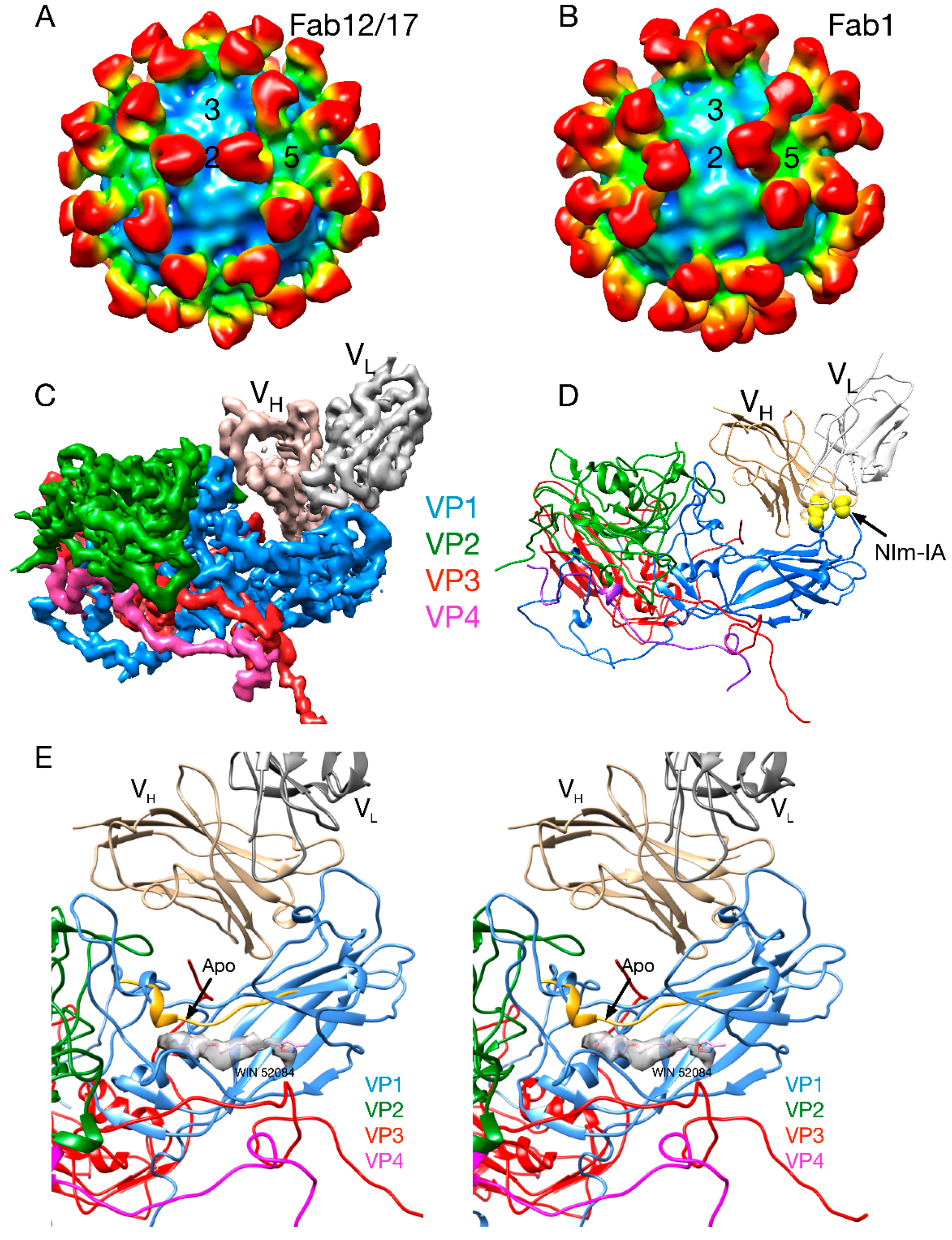
2.2.1. Structures of HRV14/Antibody Complexes
2.2.2. Antibodies to the Buried N-Termini Involved in Breathing
2.2.3. Do Antibodies Induce Conformational Changes in the Capsid?
3. Mouse Norovirus (MNV)
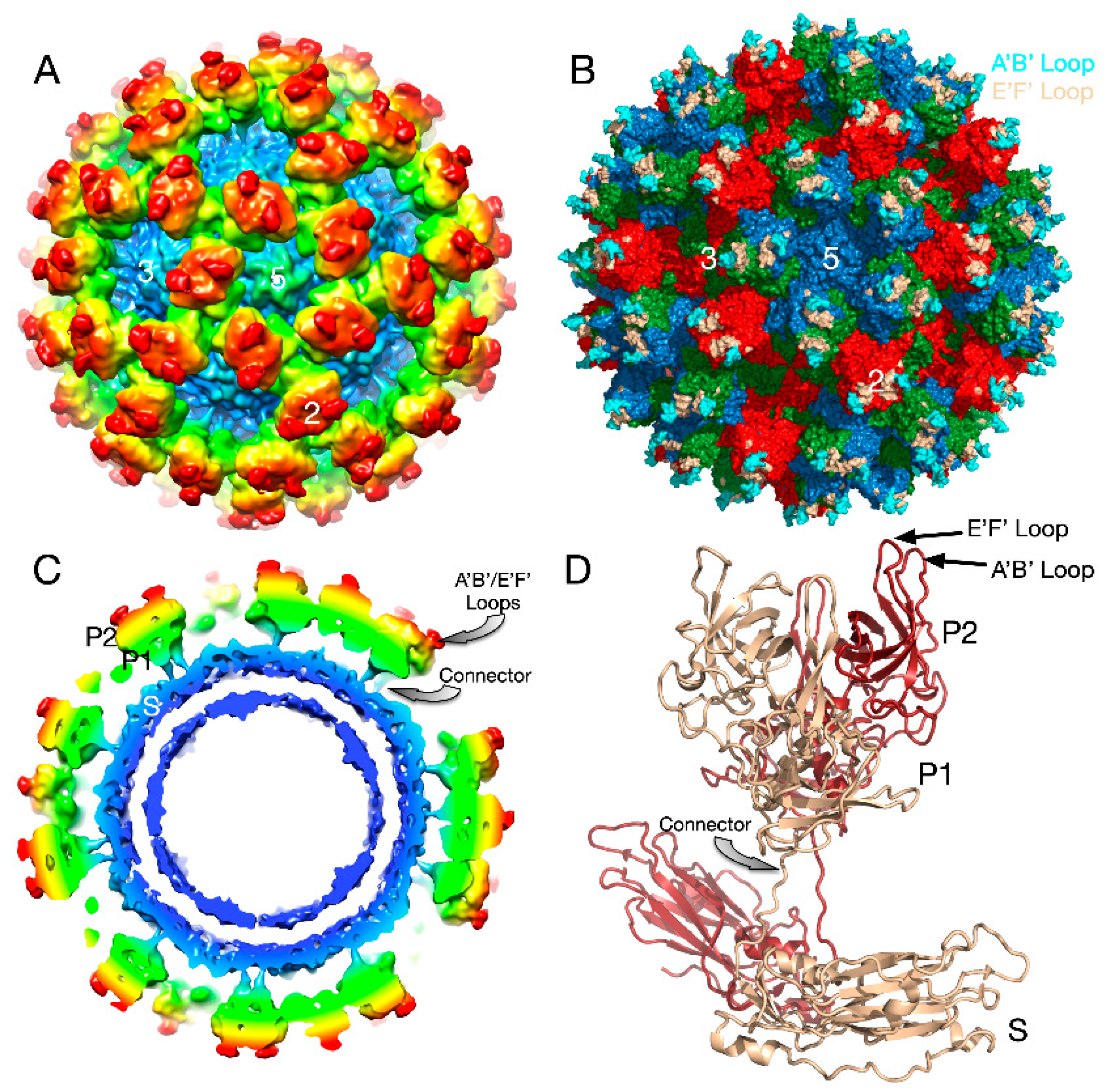
3.1. The First Mode of Flexibility; “Floating” P Domains
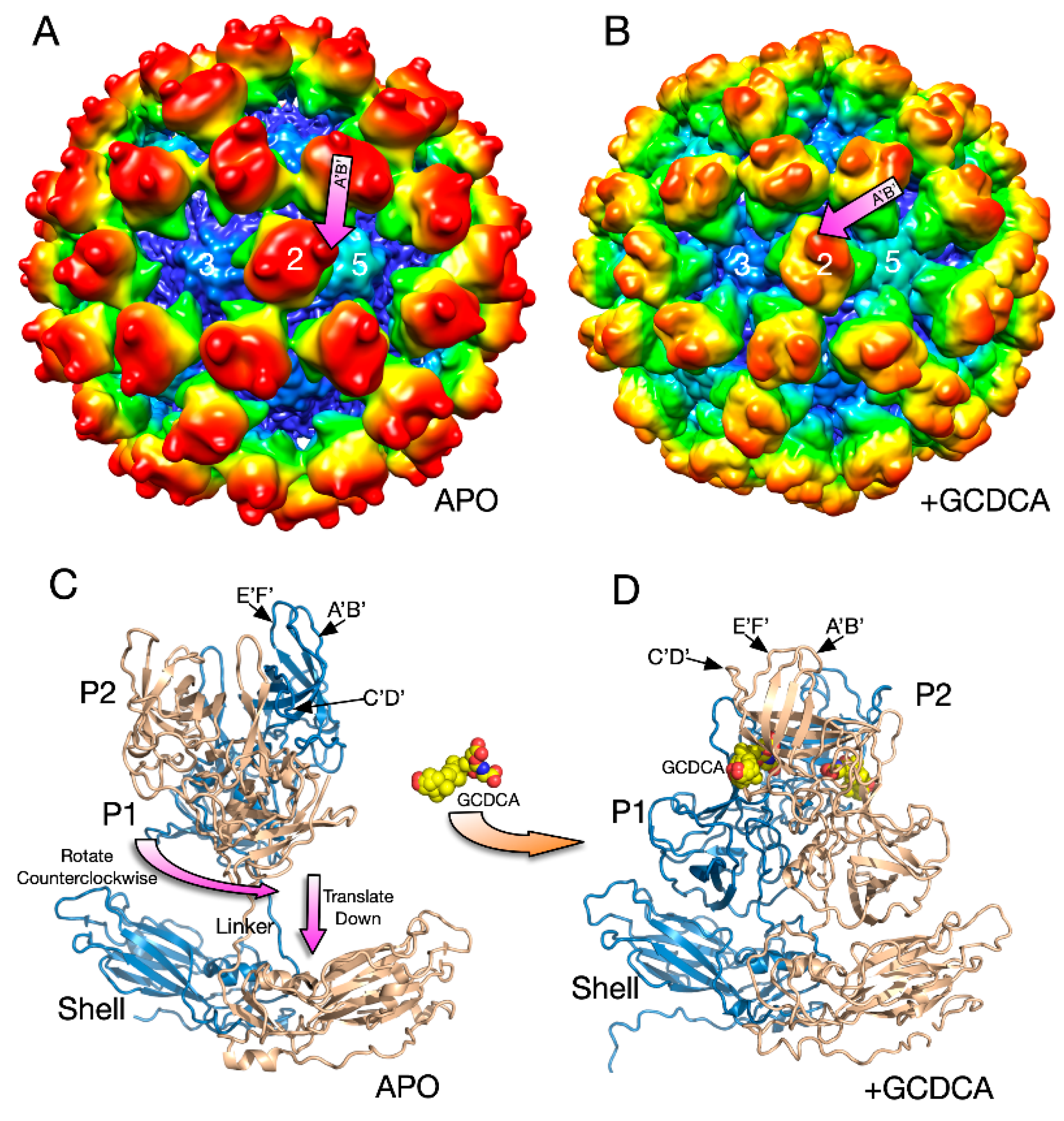
3.2. MNV–Receptor Interactions
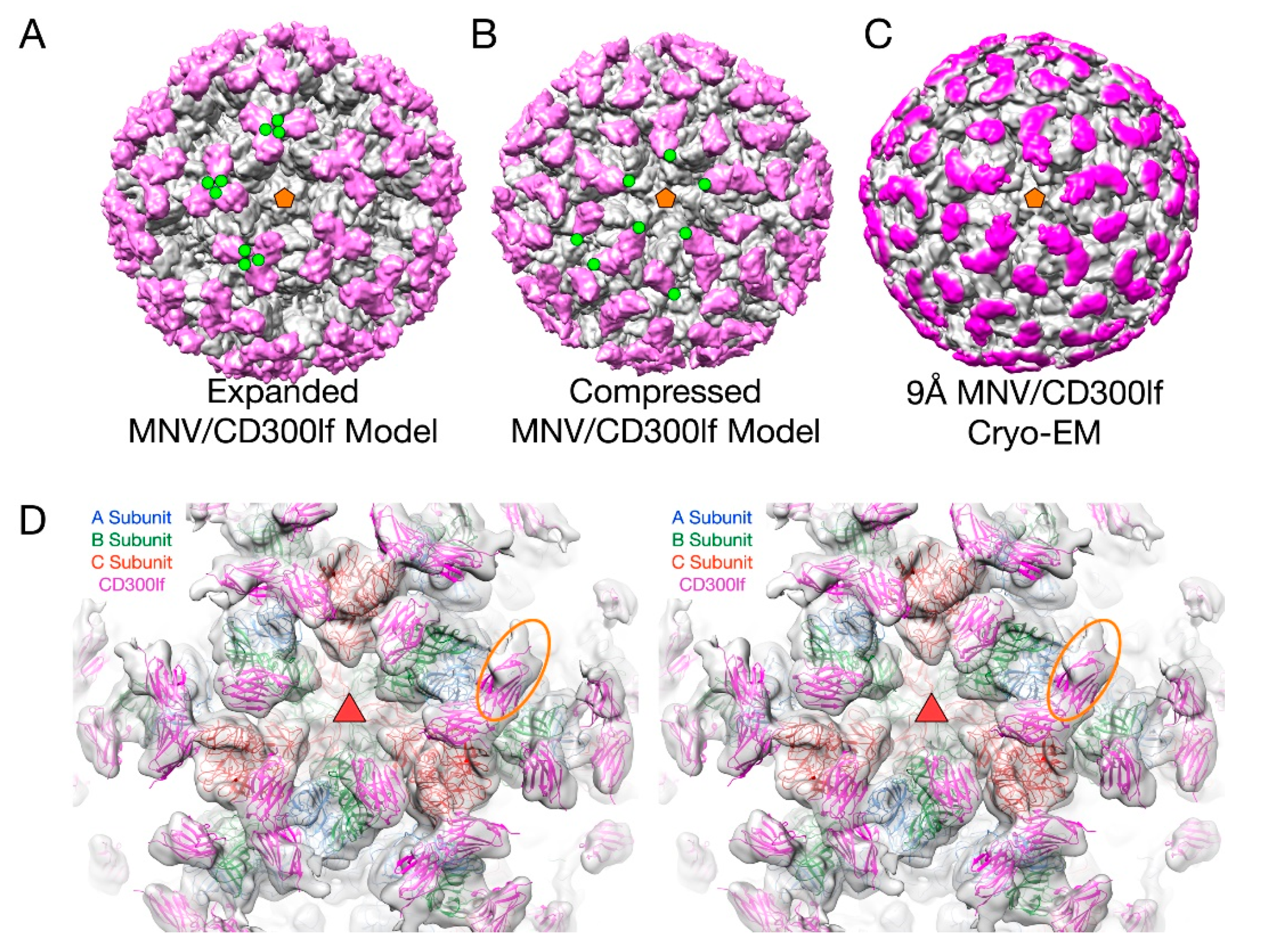
3.3. The Second Mode of Flexibility; within the P Domains
3.4. MNV P Domain Flexibility and Antibody Escape
3.5. MNV–Receptor Interactions
4. Summary
Funding
Conflicts of Interest
References
- Lewis, J.K.; Bothner, B.; Smith, T.J.; Siuzdak, G. Antiviral agent blocks breathing of the common cold virus. Proc. Natl. Acad. Sci. USA 1998, 95, 6774–6778. [Google Scholar] [CrossRef] [PubMed]
- Sherman, M.B.; Williams, A.N.; Smith, H.Q.; Nelson, C.; Wilen, C.B.; Fremont, D.H.; Virgin, H.W.; Smith, T.J. Bile Salts Alter the Mouse Norovirus Capsid Conformation: Possible Implications for Cell Attachment and Immune Evasion. J. Virol. 2019, 93, e00970-19. [Google Scholar] [CrossRef] [PubMed]
- Rueckert, R.R. Picornaviridae and Their Replication. In Fundamental Virology; Fields, B.N., Knipe, D.M., Eds.; Raven Press: New York, NY, USA, 1996; pp. 609–654. [Google Scholar]
- Rossmann, M.G.; Arnold, E.; Erickson, J.W.; Frankenberger, E.A.; Griffith, J.P.; Hecht, H.-J.; Johnson, J.E.; Kamer, G.; Luo, M.; Mosser, A.G.; et al. Structure of a human common cold virus and functional relationship to other picornaviruses. Nature 1985, 317, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Colonno, R.J.; Condra, J.H.; Mizutani, S.; Callahan, P.L.; Davies, M.E.; Murcko, M.A. Evidence for the direct involvement of the rhinovirus canyon in receptor binding. Proc. Natl. Acad. Sci. USA 1988, 85, 5449–5453. [Google Scholar] [CrossRef]
- Kolatkar, P.; Bella, J.; Olson, N.H.; Bator, C.M.; Baker, T.S.; Rossmann, M.G. Structural studies of two rhinovirus serotypes complexed with fragments of their cellular receptor. EMBO J. 1999, 18, 6249–6259. [Google Scholar] [CrossRef]
- Olson, N.H.; Kolatkar, P.; Oliveira, M.A.; Greve, J.M.; McClelland, A.; Baker, T.S.; Cheng, R.H.; Rossmann, M.G. Structure of a human Rhinovirus complexed with its receptor molecule. Protein Eng. Des. Sel. 1993, 90, 507–511. [Google Scholar]
- Sherry, B.; Mosser, A.G.; Colonno, R.J.; Rueckert, R.R. Use of monoclonal antibodies to identify four neutralization immunogens on a common cold picornavirus, human rhinovirus 14. J. Virol. 1986, 57, 246–257. [Google Scholar] [CrossRef]
- Sherry, B.; Rueckert, R. Evidence for at least two dominant neutralization antigens on human rhinovirus 14. J. Virol. 1985, 53, 137–143. [Google Scholar] [CrossRef]
- Smith, T.J.; Kremer, M.; Luo, M.; Vriend, G.; Arnold, E.; Kamer, G.; Rossmann, M.; McKinlay, M.; Diana, G.; Otto, M. The site of attachment in human rhinovirus 14 for antiviral agents that inhibit uncoating. Science 1986, 233, 1286–1293. [Google Scholar] [CrossRef]
- Fox, M.P.; Otto, M.J.; McKinlay, M.A. Prevention of rhinovirus and poliovirus uncoating by WIN 51711, a new antiviral drug. Antimicrob. Agents Chemother. 1986, 30, 110–116. [Google Scholar] [CrossRef]
- Badger, J.; Minor, I.; Kremer, M.J.; Oliveira, M.A.; Smith, T.J.; Griffith, J.P.; Guérin, D.M.A.; Krishnaswamy, S.; Luo, M.; Rossmann, M.G. Structural analysis of a series of antiviral agents complexed with human rhinovirus 14. Proc. Natl. Acad. Sci. USA 1988, 85, 3304–3308. [Google Scholar] [CrossRef] [PubMed]
- Badger, J.; Minor, I.; Oliveira, M.A.; Smith, T.J.; Rossmann, M.G. Structural analysis of antiviral agents that interact with the capsid of human rhinoviruses. Proteins Struct. Funct. Bioinform. 1989, 6, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, A.C.; Ishimaru, D.; Gonçalves, R.B.; Smith, T.J.; Mason, P.; Sá-Carvalho, D.; Silva, J.L. Low Temperature and Pressure Stability of Picornaviruses: Implications for Virus Uncoating. Biophys. J. 1999, 76, 1270–1279. [Google Scholar] [CrossRef]
- Pevear, D.C.; Fancher, M.J.; Felock, P.J.; Rossmann, M.G.; Miller, M.S.; Diana, G.; Treasurywala, A.M.; McKinlay, M.A.; Dutko, F.J. Conformational change in the floor of the human rhinovirus canyon blocks adsorption to HeLa cell receptors. J. Virol. 1989, 63, 2002–2007. [Google Scholar] [CrossRef]
- Marlovits, T.C.; Abrahamsberg, C.; Blaas, D. Soluble LDL minireceptors. Minimal structure requirements for recognition of minor group human rhinovirus. J. Boil. Chem. 1998, 273, 33835–33840. [Google Scholar] [CrossRef]
- Marlovits, T.; Zechmeister, T.; Gruenberger, M.; Ronacher, B.; Schwihla, H.; Blaas, D. Recombinant soluble low density lipoprotein receptor fragment inhibits minor group rhinovirus infection in vitro. FASEB J. 1998, 12, 695–703. [Google Scholar] [CrossRef]
- Hewat, E.; Neumann, E.; Conway, J.F.; Moser, R.; Ronacher, B.; Marlovits, T.; Blaas, D. The cellular receptor to human rhinovirus 2 binds around the 5-fold axis and not in the canyon: A structural view. EMBO J. 2000, 19, 6317–6325. [Google Scholar] [CrossRef]
- Hoover-Litty, H.; Greve, J.M. Formation of rhinovirus-soluble ICAM-1 complexes and conformational changes in the virion. J. Virol. 1993, 67, 390–397. [Google Scholar] [CrossRef]
- Oliveira, M.A.; Zhao, R.; Lee, W.-M.; Kremer, M.J.; Minor, I.; Rueckert, R.R.; Diana, G.D.; Pevear, D.C.; Dutko, F.J.; McKinlay, M.A.; et al. The structure of human rhinovirus 16. Structure 1993, 1, 51–68. [Google Scholar] [CrossRef]
- Smith, T.J.; Chase, E.S.; Schmidt, T.J.; Olson, N.H.; Baker, T.S. Neutralizing antibody to human rhinovirus 14 penetrates the receptor-binding canyon. Nature 1996, 383, 350–354. [Google Scholar] [CrossRef]
- Katpally, U.; Smith, T.J. Pocket Factors Are Unlikely To Play a Major Role in the Life Cycle of Human Rhinovirus. J. Virol. 2007, 81, 6307–6315. [Google Scholar] [CrossRef]
- Matthews, B.W. Structural and genetic analysis of protein folding and stability. Curr. Opin. Struct. Boil. 1993, 3, 589–593. [Google Scholar] [CrossRef]
- Smith, T.J.; Baker, T. Picornaviruses: Epitopes, Canyons, and Pockets. Adv. Appl. Microbiol. 1999, 52, 1–23. [Google Scholar]
- Holland, J.J. Irreversible eclipse of poliovirus by HeLa cells. Virology 1962, 16, 163–176. [Google Scholar] [CrossRef]
- Holland, J.J.; Hoyer, B.H. Early Stages of Enterovirus Infection. Cold Spring Harb. Symp. Quant. Boil. 1962, 27, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Liu, Y.; Jiang, W.; Smith, T.J.; Xu, Z.; Rossmann, M.G. Antibody-induced uncoating of human rhinovirus B14. Proc. Natl. Acad. Sci. USA 2017, 114, 8017–8022. [Google Scholar] [CrossRef]
- Dulbecco, R.; Vogt, M.; Strickland, A. A study of the basic aspects of neutralization of two animal viruses, Western equine encephalitis virus and poliomyelitis virus. Virology 1956, 2, 162–205. [Google Scholar] [CrossRef]
- Emini, E.A.; Kao, S.Y.; Lewis, A.J.; Crainic, R.; Wimmer, E. Functional basis of poliovirus neutralization determined with monospecific neutralizing antibodies. J. Virol. 1983, 46, 466–474. [Google Scholar] [CrossRef]
- Emini, E.A.; Ostapchuk, P.; Wimmer, E. Bivalent attachment of antibody onto poliovirus leads to conformational alteration and neutralization. J. Virol. 1983, 48, 547–550. [Google Scholar] [CrossRef]
- Mandel, B. Neutralization of poliovirus: A hypothesis to explain the mechanism and the one-hit character of the neutralization reaction. Virology 1976, 69, 500–510. [Google Scholar] [CrossRef]
- Wetz, K.; Willingmann, P.; Zeichhardt, H.; Habermehl, K.-O. Neutralization of poliovirus by polyclonal antibodies requires binding of a single IgG molecule per virion. Arch. Virol. 1986, 91, 207–220. [Google Scholar] [CrossRef] [PubMed]
- Colonno, R.J.; Callahan, P.L.; Leippe, D.M.; Rueckert, R.R.; Tomassini, J.E. Inhibition of rhinovirus attachment by neutralizing monoclonal antibodies and their Fab fragments. J. Virol. 1989, 63, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Vrijsen, R.; Mosser, A.; Boeye, A. Post-absorption neutralization of poliovirus. J. Virol. 1993, 67, 3126–3133. [Google Scholar] [CrossRef] [PubMed]
- Mosser, A.G.; Leippe, D.M.; Rueckert, R.R. Neutralization of picornaviruses: Support for the pentamer bridging hypothesis. In Molecular Aspects of Picornavirus Infection and Detection; Semler, B.L., Ehrenfeld, E., Eds.; American Society for Microbiology: Washington, DC, USA, 1989; pp. 155–167. [Google Scholar]
- Leippe, D.M. Stoichiometry of Picornavirus Neutralization by Murine Monoclonal Antibodies; University of Wisconsin: Madison, WI, USA, 1991. [Google Scholar]
- Smith, T.J.; Olson, N.H.; Cheng, R.H.; Chase, E.S.; Baker, T.S. Structure of a human rhinovirus-bivalently bound antibody complex: Implications for viral neutralization and antibody flexibility. Proc. Natl. Acad. Sci. USA 1993, 90, 7015–7018. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.J.; Olson, N.H.; Cheng, R.H.; Liu, H.; Chase, E.S.; Lee, W.M.; Leippe, D.M.; Mosser, A.G.; Rueckert, R.R.; Baker, T.S. Structure of human rhinovirus complexed with Fab fragments from a neutralizing antibody. J. Virol. 1993, 67, 1148–1158. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Smith, T.J.; Lee, W.-M.; Mosser, A.G.; Rueckert, R.R.; Olson, N.H.; Cheng, R.; Baker, T.S. Structure Determination of an Fab Fragment that Neutralizes Human Rhinovirus 14 and Analysis of the Fab-Virus Complex. J. Mol. Boil. 1994, 240, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Che, Z.; Olson, N.H.; Leippe, D.; Lee, W.-M.; Mosser, A.; Rueckert, R.R.; Baker, T.S.; Smith, T.J. Antibody-mediated neutralization of human rhinovirus 14 explored by means of cryo-electron microscopy and X-ray crystallography of virus-Fab complexes. J. Virol. 1998, 72, 4610–4622. [Google Scholar] [CrossRef]
- Katpally, U.; Fu, T.-M.; Freed, D.C.; Casimiro, D.R.; Smith, T.J. Antibodies to the Buried N Terminus of Rhinovirus VP4 Exhibit Cross-Serotypic Neutralization. J. Virol. 2009, 83, 7040–7048. [Google Scholar] [CrossRef]
- Smith, T.J.; Mosser, A.G. Antibody-mediated neutralization of picornaviruses. In Structure Biology of Viruses; Chiu, W., Burnett, R., Garcea, R., Eds.; Oxford University Press: New York, NY, USA, 1995. [Google Scholar]
- Smith, T.J.; Mosser, A.G.; Baker, T.S. Structural studies on the mechanisms of antibody-mediated neutralization of human rhinovirus. Semin. Virol. 1995, 6, 233–242. [Google Scholar] [CrossRef]
- Hutson, A.M.; Atmar, R.L.; Estes, M.K. Norovirus disease: Changing epidemiology and host susceptibility factors. Trends Microbiol. 2004, 12, 279–287. [Google Scholar] [CrossRef]
- Moe, C.L.; Sobsey, M.D.; Stewart, P.W.; Crawford-Brown, D. Estimating the risk of human calicivirus infection from drinking water. In Proceedings of the International Workshop on Human Caliciviruses, Atlanta, GA, USA, 29–31 March 1999; pp. 4–6. [Google Scholar]
- Blanton, L.H.; Adams, S.M.; Beard, R.S.; Wei, G.; Bulens, S.N.; Widdowson, M.-A.; Glass, R.I.; Monroe, S.S. Molecular and Epidemiologic Trends of Caliciviruses Associated with Outbreaks of Acute Gastroenteritis in the United States, 2000–2004. J. Infect. Dis. 2006, 193, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Siebenga, J.J.; Vennema, H.; Duizer, E.; Koopmans, M.P. Gastroenteritis Caused by Norovirus GGII.4, the Netherlands, 1994–2005. Emerg. Infect. Dis. 2007, 13, 144–146. [Google Scholar] [CrossRef] [PubMed]
- Hall, A.J.; Lopman, B.A.; Payne, D.C.; Patel, M.M.; Gastañaduy, P.A.; Vinjé, J.; Parashar, U.D. Norovirus Disease in the United States. Emerg. Infect. Dis. 2013, 19, 1198–1205. [Google Scholar] [CrossRef] [PubMed]
- Debbink, K.; Lindesmith, L.C.; Donaldson, E.F.; Costantini, V.; Beltramello, M.; Corti, D.; Swanstrom, J.; Lanzavecchia, A.; Vinjé, J.; Baric, R.S. Emergence of new pandemic GII.4 Sydney norovirus strain correlates with escape from herd immunity. J. Infect. Dis. 2013, 208, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Ettayebi, K.; Crawford, S.E.; Murakami, K.; Broughman, J.R.; Karandikar, U.; Tenge, V.R.; Neill, F.H.; Blutt, S.E.; Zeng, X.-L.; Qu, L.; et al. Replication of human noroviruses in stem cell-derived human enteroids. Science 2016, 353, 1387–1393. [Google Scholar] [CrossRef] [PubMed]
- Costantini, V.; Morantz, E.K.; Browne, H.; Ettayebi, K.; Zeng, X.-L.; Atmar, R.L.; Estes, M.K.; Vinjé, J. Human Norovirus Replication in Human Intestinal Enteroids as Model to Evaluate Virus Inactivation. Emerg. Infect. Dis. 2018, 24, 1453–1464. [Google Scholar] [CrossRef] [PubMed]
- Taube, S.; Kolawole, A.O.; Höhne, M.; Wilkinson, J.E.; Handley, S.A.; Perry, J.W.; Thackray, L.B.; Akkina, R.; Wobus, C.E. A Mouse Model for Human Norovirus. mBio 2013, 4, e00450-13. [Google Scholar] [CrossRef]
- Prasad, B.V.V.; Hardy, M.E.; Dokland, T.; Bella, J.; Rossmann, M.G.; Estes, M.K. X-ray Crystallographic Structure of the Norwalk Virus Capsid. Science 1999, 286, 287–290. [Google Scholar] [CrossRef]
- Prasad, B.V.V.; Hardy, M.E.; Jiang, X.; Estes, M.K. Structure of Norwalk virus. Arch. Virol. Suppl. 1996, 12, 237–242. [Google Scholar]
- Prasad, B.V.V.; Matson, D.; Smith, A. Three-dimensional Structure of Calicivirus. J. Mol. Boil. 1994, 240, 256–264. [Google Scholar] [CrossRef]
- Katpally, U.; Wobus, C.E.; Dryden, K.; Virgin, H.W.; Smith, T.J. Structure of Antibody-Neutralized Murine Norovirus and Unexpected Differences from Viruslike Particles. J. Virol. 2007, 82, 2079–2088. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.-M.; Hutson, A.M.; Estes, M.K.; Prasad, B.V.V. Atomic resolution structural characterization of recognition of histo-blood group antigens by Norwalk virus. Proc. Natl. Acad. Sci. USA 2008, 105, 9175–9180. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.; Hegde, R.S.; Jiang, X. The P Domain of Norovirus Capsid Protein Forms Dimer and Binds to Histo-Blood Group Antigen Receptors. J. Virol. 2004, 78, 6233–6242. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, E.F.; Lindesmith, L.C.; LoBue, A.D.; Baric, R.S. Viral shape-shifting: Norovirus evasion of the human immune system. Nat. Rev. Genet. 2010, 8, 231–241. [Google Scholar] [CrossRef]
- Nilsson, M.; Hedlund, K.-O.; Thorhagen, M.; Larson, G.; Johansen, K.; Ekspong, A.; Svensson, L. Evolution of Human Calicivirus RNA In Vivo:Accumulation of Mutations in the Protruding P2 Domain of the CapsidLeads to Structural Changes and Possibly a NewPhenotype. J. Virol. 2003, 77, 13117–13124. [Google Scholar] [CrossRef]
- Taube, S.; Rubin, J.R.; Katpally, U.; Smith, T.J.; Kendall, A.; Stuckey, J.A.; Wobus, C.E. High-Resolution X-ray Structure and Functional Analysis of the Murine Norovirus 1 Capsid Protein Protruding Domain. J. Virol. 2010, 84, 5695–5705. [Google Scholar] [CrossRef]
- Hansman, G.S.; Taylor, D.W.; McLellan, J.S.; Smith, T.J.; Georgiev, I.; Tame, J.R.H.; Park, S.-Y.; Yamazaki, M.; Gondaira, F.; Miki, M.; et al. Structural Basis for Broad Detection of Genogroup II Noroviruses by a Monoclonal Antibody That Binds to a Site Occluded in the Viral Particle. J. Virol. 2012, 86, 3635–3646. [Google Scholar] [CrossRef]
- Ruoff, K.; Kilic, T.; Devant, J.; Koromyslova, A.; Ringel, A.; Hempelmann, A.; Geiss, C.; Graf, J.; Haas, M.; Roggenbach, I.; et al. Structural Basis of Nanobodies Targeting the Prototype Norovirus. J. Virol. 2019, 93, e02005-18. [Google Scholar] [CrossRef]
- Lindesmith, L.C.; Mallory, M.L.; Debbink, K.; Donaldson, E.F.; Brewer-Jensen, P.D.; Swann, E.W.; Sheahan, T.P.; Graham, R.L.; Beltramello, M.; Corti, D.; et al. Conformational Occlusion of Blockade Antibody Epitopes, a Novel Mechanism of GII.4 Human Norovirus Immune Evasion. mSphere 2018, 3, e00518-17. [Google Scholar] [CrossRef]
- Orchard, R.C.; Wilen, C.B.; Doench, J.G.; Baldridge, M.T.; McCune, B.T.; Lee, Y.-C.; Lee, S.; Pruett-Miller, S.M.; Nelson, C.A.; Fremont, D.H.; et al. Discovery of a proteinaceous cellular receptor for a norovirus. Science 2016, 353, 933–936. [Google Scholar] [CrossRef]
- Nelson, C.A.; Wilen, C.B.; Dai, Y.-N.; Orchard, R.C.; Kim, A.S.; Stegeman, R.A.; Hsieh, L.L.; Smith, T.J.; Virgin, H.W.; Fremont, D.H. Structural basis for murine norovirus engagement of bile acids and the CD300lf receptor. Proc. Natl. Acad. Sci. USA 2018, 115, E9201. [Google Scholar] [CrossRef] [PubMed]
- Hornick, C.L.; Karush, F. Antibody affinity-III. The role of multivalence. Immunochemistry 1972, 9, 325–340. [Google Scholar] [CrossRef]
- Kilic, T.; Koromyslova, A.; Malak, V.; Hansman, G.S. Atomic Structure of the Murine Norovirus Protruding Domain and Soluble CD300lf Receptor Complex. J. Virol. 2018, 92, e00413-18. [Google Scholar] [CrossRef] [PubMed]
- Kolawole, A.O.; Xia, C.; Li, M.; Gamez, M.; Yu, C.; Rippinger, C.M.; Yucha, R.E.; Smith, T.J.; Wobus, C.E. Newly isolated mAbs broaden the neutralizing epitope in murine norovirus. J. Gen. Virol. 2014, 95, 1958–1968. [Google Scholar] [CrossRef]
- Kolawole, A.O.; Li, M.; Xia, C.; Fischer, A.E.; Giacobbi, N.; Rippinger, C.M.; Proescher, J.B.G.; Wu, S.K.; Bessling, S.L.; Gamez, M.; et al. Flexibility in Surface-Exposed Loops in a Virus Capsid Mediates Escape from Antibody Neutralization. J. Virol. 2014, 88, 4543–4557. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kolawole, A.O.; Smith, H.Q.; Svoboda, S.A.; Lewis, M.S.; Sherman, M.B.; Lynch, G.C.; Pettitt, B.M.; Smith, T.J.; Wobus, C.E. Norovirus escape from broadly neutralizing antibodies is limited to allosteric-like mechanisms. mSphere 2017, 2, e00334-17. [Google Scholar] [CrossRef]
- Parry, N.; Fox, G.; Rowlands, D.; Brown, F.; Fry, E.E.; Acharya, R.; Logan, D.T.; Stuart, D.I. Structural and serological evidence for a novel mechanism of antigenic variation in foot-and-mouth disease virus. Nature 1990, 347, 569–572. [Google Scholar] [CrossRef]
- Verdaguer, N.; Schoehn, G.; Ochoa, W.F.; Fita, I.; Brookes, S.M.; King, A.; Domingo, E.; Mateu, M.G.; Stuart, D.I.; Hewat, E. Flexibility of the Major Antigenic Loop of Foot-and-Mouth Disease Virus Bound to a Fab Fragment of a Neutralising Antibody: Structure and Neutralisation. Virology 1999, 255, 260–268. [Google Scholar] [CrossRef]
- Ma, S.; Lavin, B. SPC update: Advocate General opines on the meaning of ‘protected by’. Pharm. Pat. Anal. 2018, 7, 137–140. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sherman, M.B.; Smith, H.Q.; Smith, T.J. The Dynamic Life of Virus Capsids. Viruses 2020, 12, 618. https://doi.org/10.3390/v12060618
Sherman MB, Smith HQ, Smith TJ. The Dynamic Life of Virus Capsids. Viruses. 2020; 12(6):618. https://doi.org/10.3390/v12060618
Chicago/Turabian StyleSherman, Michael B., Hong Q. Smith, and Thomas J. Smith. 2020. "The Dynamic Life of Virus Capsids" Viruses 12, no. 6: 618. https://doi.org/10.3390/v12060618
APA StyleSherman, M. B., Smith, H. Q., & Smith, T. J. (2020). The Dynamic Life of Virus Capsids. Viruses, 12(6), 618. https://doi.org/10.3390/v12060618