Discovery of Three Toxic Proteins of Klebsiella Phage fHe-Kpn01

 ,
,  and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Bacterial Strains, Phage, and Media
2.2. Phage Isolation and Purification
2.3. Electron Microscopy
2.4. Host Range Determination
2.5. Infection Growth Curves
2.6. Genome Sequencing and Analysis
2.7. Restriction Fragment Analysis of Phage DNA
2.8. Detection of Nicks in the Phage DNA
2.9. Phylogenetic Analysis
2.10. Initial Screening of HPUFs for Toxicity
2.11. Confirmation of Protein Toxicity
2.12. Prediction of the Structure of Toxic Proteins
3. Results
3.1. General Genomic Features of fHe-Kpn01
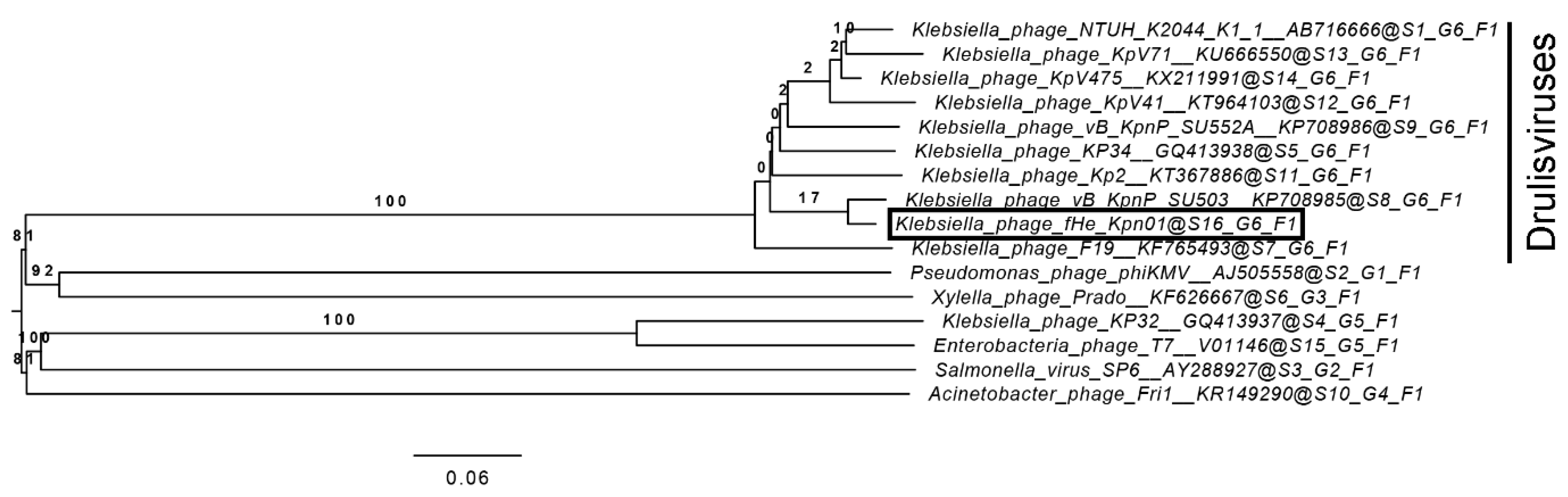
3.2. Comparison of fHe-Kpn01 to Closely Related Phages
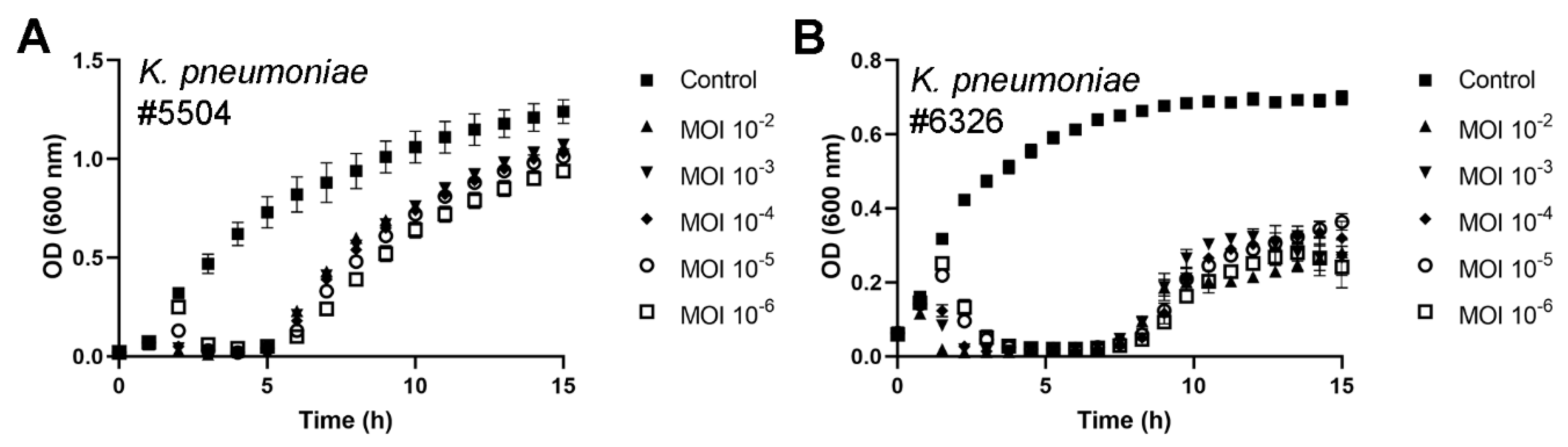
3.3. fHe-Kpn01 Host Range and Growth Characteristics
3.4. Antibacterial Potential of fHe-Kpn01
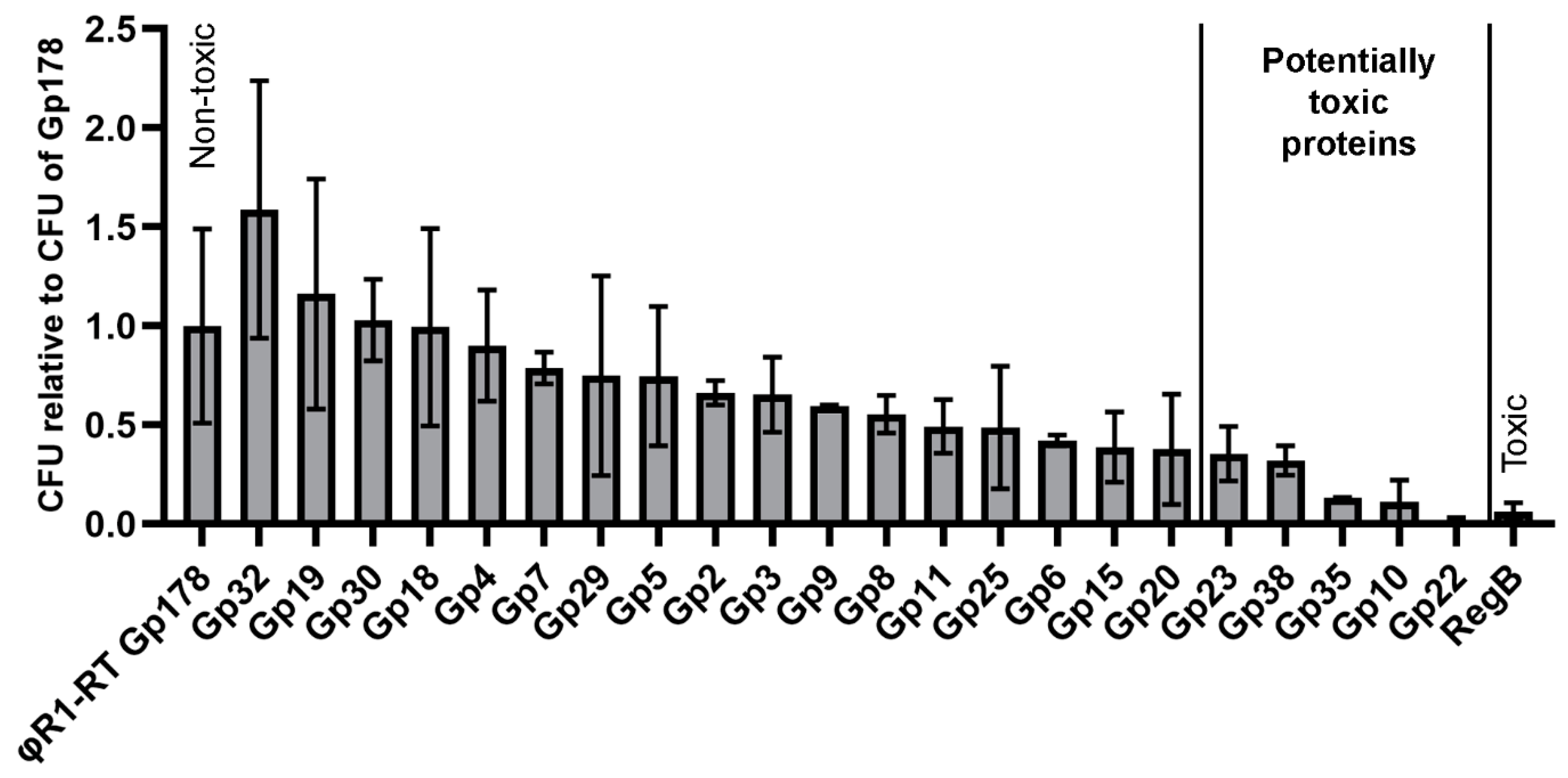
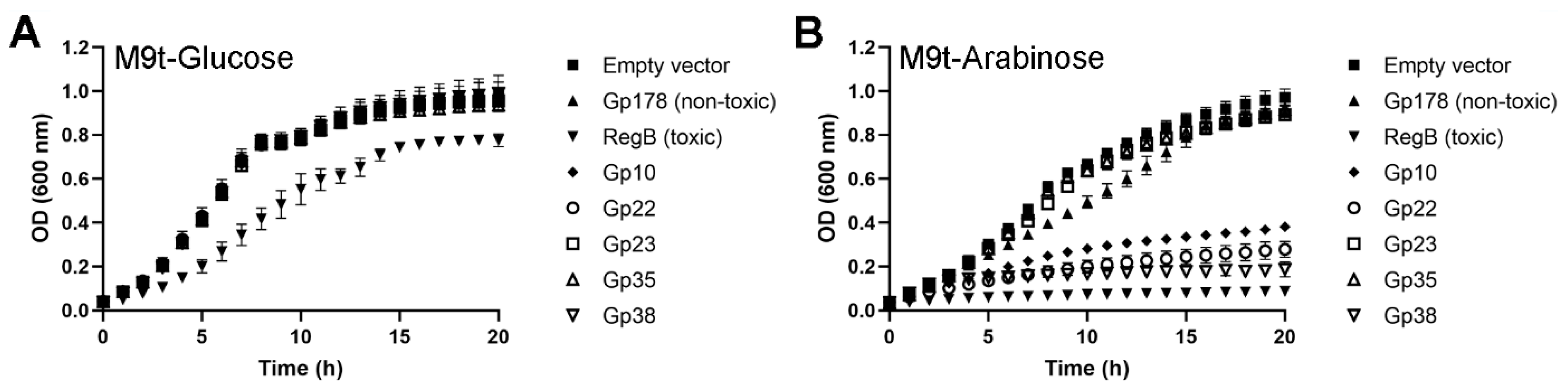
3.5. Three Toxic Proteins are Encoded in the Genome of fHe-Kpn01
3.6. The Structure of Gp22 Resembles Nucleotidyltransferases
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tagliabue, A.; Rappuoli, R. Changing priorities in vaccinology: Antibiotic resistance moving to the top. Front. Immunol. 2018, 9, 1068. [Google Scholar] [CrossRef] [PubMed]
- Navon-Venezia, S.; Kondratyeva, K.; Carattoli, A. Klebsiella pneumoniae: A major worldwide source and shuttle for antibiotic resistance. FEMS Microbiol. Rev. 2017, 41, 252–275. [Google Scholar] [CrossRef] [PubMed]
- Giske, C.G.; Monnet, D.L.; Cars, O.; Carmeli, Y.; ReAct-Action on Antibiotic. Clinical and economic impact of common multidrug-resistant gram-negative bacilli. Antimicrob. Agents Chemother. 2008, 52, 813–821. [Google Scholar] [CrossRef] [PubMed]
- Hersh, A.L.; Newland, J.G.; Beekmann, S.E.; Polgreen, P.M.; Gilbert, D.N. Unmet medical need in infectious diseases. Clin. Infect. Dis. 2012, 54, 1677–1678. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO publishes list of bacteria for which new antibiotics are urgently needed. Available online: https://www.who.int/en/news-room/detail/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed on 9 June 2019).
- Gordillo Altamirano, F.L.; Barr, J.J. Phage therapy in the postantibiotic era. Clin. Microbiol. Rev. 2019, 32, e00066-18. [Google Scholar] [CrossRef] [PubMed]
- Hesse, S.; Adhya, S. Phage therapy in the twenty-first century: Facing the decline of the antibiotic era; Is it finally time for the age of the phage? Annu. Rev. Microbiol. 2019, 73, 155–174. [Google Scholar] [CrossRef] [PubMed]
- Roach, D.R.; Donovan, D.M. Antimicrobial bacteriophage-derived proteins and therapeutic applications. Bacteriophage 2015, 5, e1062590. [Google Scholar] [CrossRef]
- Roucourt, B.; Lavigne, R. The role of interactions between phage and bacterial proteins within the infected cell: A diverse and puzzling interactome. Environ. Microbiol. 2009, 11, 2789–2805. [Google Scholar] [CrossRef]
- Pope, W.H.; Bowman, C.A.; Russell, D.A.; Jacobs-Sera, D.; Asai, D.J.; Cresawn, S.G.; Jacobs, W.R.; Hendrix, R.W.; Lawrence, J.G.; Hatfull, G.F.; et al. Whole genome comparison of a large collection of mycobacteriophages reveals a continuum of phage genetic diversity. Elife 2015, 4, e06416. [Google Scholar] [CrossRef]
- Lorenz, L.; Lins, B.; Barrett, J.; Montgomery, A.; Trapani, S.; Schindler, A.; Christie, G.E.; Cresawn, S.G.; Temple, L. Genomic characterization of six novel Bacillus pumilus bacteriophages. Virology 2013, 444, 374–383. [Google Scholar] [CrossRef]
- Seguritan, V.; Feng, I.W.; Rohwer, F.; Swift, M.; Segall, A.M. Genome sequences of two closely related Vibrio parahaemolyticus phages, VP16T and VP16C. J. Bacteriol. 2003, 185, 6434–6447. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Dehbi, M.; Moeck, G.; Arhin, F.; Bauda, P.; Bergeron, D.; Callejo, M.; Ferretti, V.; Ha, N.; Kwan, T.; et al. Antimicrobial drug discovery through bacteriophage genomics. Nat. Biotechnol. 2004, 22, 185–191. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Godavarthi, S.; Kumar, A.; Sen, R. A mycobacteriophage genomics approach to identify novel mycobacteriophage proteins with mycobactericidal properties. Microbiology 2019, 165, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Van den Bossche, A.; Ceyssens, P.J.; De Smet, J.; Hendrix, H.; Bellon, H.; Leimer, N.; Wagemans, J.; Delattre, A.S.; Cenens, W.; Aertsen, A.; et al. Systematic identification of hypothetical bacteriophage proteins targeting key protein complexes of Pseudomonas aeruginosa. J. Proteome Res. 2014, 13, 4446–4456. [Google Scholar] [CrossRef] [PubMed]
- Shibayama, Y.; Dabbs, E.R. Phage as a source of antibacterial genes: Multiple inhibitory products encoded by Rhodococcus phage YF1. Bacteriophage 2011, 1, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Mohanraj, U.; Wan, X.; Spruit, C.M.; Skurnik, M.; Pajunen, M.I. A toxicity screening approach to identify bacteriophage-encoded anti-microbial proteins. Viruses 2019, 11, 1057. [Google Scholar] [CrossRef]
- Grant, S.G.; Jessee, J.; Bloom, F.R.; Hanahan, D. Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc. Natl. Acad. Sci. USA 1990, 87, 4645–4649. [Google Scholar] [CrossRef]
- Hanahan, D. Studies on transformation of Escherichia coli with plasmids. J. Mol. Biol. 1983, 166, 557–580. [Google Scholar] [CrossRef]
- Lamberg, A.; Nieminen, S.; Qiao, M.; Savilahti, H. Efficient insertion mutagenesis strategy for bacterial genomes involving electroporation of in vitro-assembled DNA transposition complexes of bacteriophage Mu. Appl. Environ. Microbiol. 2002, 68, 705–712. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.; Maniatis, T.J. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; pp. 9.14–19.19. [Google Scholar]
- Ackermann, H.W. Basic phage electron microscopy. In Bacteriophages Methods and Protocols; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press Inc.: Totowa, NJ, USA, 2009; volume 2, pp. 113–126. [Google Scholar]
- Coil, D.; Jospin, G.; Darling, A.E. A5-miseq: An updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 2015, 31, 587–589. [Google Scholar] [CrossRef]
- Garneau, J.R.; Depardieu, F.; Fortier, L.C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef] [PubMed]
- Salem, M.; Skurnik, M. Genomic characterization of sixteen Yersinia enterocolitica-infecting podoviruses of pig origin. Viruses 2018, 10, 174. [Google Scholar] [CrossRef] [PubMed]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, L.; Stephens, A.; Nam, S.Z.; Rau, D.; Kubler, J.; Lozajic, M.; Gabler, F.; Soding, J.; Lupas, A.N.; Alva, V. A completely reimplemented MPI bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE on-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Voldby Larsen, M. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Joensen, K.G.; Scheutz, F.; Lund, O.; Hasman, H.; Kaas, R.S.; Nielsen, E.M.; Aarestrup, F.M. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J. Clin. Microbiol. 2014, 52, 1501–1510. [Google Scholar] [CrossRef]
- Leskinen, K.; Tuomala, H.; Wicklund, A.; Horsma-Heikkinen, J.; Kuusela, P.; Skurnik, M.; Kiljunen, S. Characterization of vB_SauM-fRuSau02, a Twort-like bacteriophage isolated from a therapeutic phage cocktail. Viruses 2017, 9, 258. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef]
- Wyres, K.L.; Wick, R.R.; Gorrie, C.; Jenney, A.; Follador, R.; Thomson, N.R.; Holt, K.E. Identification of Klebsiella capsule synthesis loci from whole genome data. Microb. Genom. 2016, 2, e000102. [Google Scholar] [CrossRef]
- Wick, R.R.; Heinz, E.; Holt, K.E.; Wyres, K.L. Kaptive Web: User-friendly capsule and lipopolysaccharide serotype prediction for Klebsiella genomes. J. Clin. Microbiol. 2018, 56, e00197-18. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses. Virus Taxonomy: 2018b Release. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 17 June 2019).
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 2013, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Meier-Kolthoff, J.P.; Göker, M. VICTOR: Genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 2017, 33, 3396–3404. [Google Scholar] [CrossRef] [PubMed]
- Lefort, V.; Desper, R.; Gascuel, O. FastME 2.0: A comprehensive, accurate, and fast distance-based phylogeny inference program. Mol. Biol. Evol. 2015, 32, 2798–2800. [Google Scholar] [CrossRef] [PubMed]
- Farris, J.S. Estimating phylogenetic trees from distance matrices. Am. Nat. 1972, 106, 645–668. [Google Scholar] [CrossRef]
- Rambaut, A. FigTree 1.4.3—A Graphical Viewer of Phylogenetic Trees and a Program for Producing Publication-Ready Figures. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 17 June 2019).
- Göker, M.; Garcia-Blazquez, G.; Voglmayr, H.; Telleria, M.T.; Martin, M.P. Molecular taxonomy of phytopathogenic fungi: A case study in Peronospora. PLoS ONE 2009, 4, e6319. [Google Scholar] [CrossRef]
- Meier-Kolthoff, J.P.; Hahnke, R.L.; Petersen, J.; Scheuner, C.; Michael, V.; Fiebig, A.; Rohde, C.; Rohde, M.; Fartmann, B.e.; Goodwin, L.A. Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genomic Sci. 2014, 9, 2. [Google Scholar] [CrossRef]
- Sanson, B.; Uzan, M. Dual role of the sequence-specific bacteriophage T4 endoribonuclease RegB. mRNA inactivation and mRNA destabilization. J. Mol. Biol. 1993, 233, 429–446. [Google Scholar] [CrossRef]
- Leon-Velarde, C.G.; Happonen, L.; Pajunen, M.; Leskinen, K.; Kropinski, A.M.; Mattinen, L.; Rajtor, M.; Zur, J.; Smith, D.; Chen, S.; et al. Yersinia enterocolitica -specific infection by bacteriophages TG1 and φR1-RT is dependent on temperature-regulated expression of the phage host receptor OmpF. Appl Environ. Microbiol 2016, 82, 5340–5353. [Google Scholar] [CrossRef]
- Guzman, L.M.; Belin, D.; Carson, M.J.; Beckwith, J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 1995, 177, 4121–4130. [Google Scholar] [CrossRef]
- Institute for Molecular Medicine Finland Technology Centre. Sequencing unit. Available online: https://www.fimm.fi/en/services/technology-centre/sequencing/ (accessed on 17 June 2019).
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, H.; Maciejewska, B.; Latka, A.; Majkowska-Skrobek, G.; Hellstrand, M.; Melefors, O.; Wang, J.T.; Kropinski, A.M.; Drulis-Kawa, Z.; Nilsson, A.S. A suggested new bacteriophage genus, “Kp34likevirus”, within the Autographivirinae subfamily of Podoviridae. Viruses 2015, 7, 1804–1822. [Google Scholar] [CrossRef] [PubMed]
- Drulis-Kawa, Z.; Mackiewicz, P.; Kesik-Szeloch, A.; Maciaszczyk-Dziubinska, E.; Weber-Dabrowska, B.; Dorotkiewicz-Jach, A.; Augustyniak, D.; Majkowska-Skrobek, G.; Bocer, T.; Empel, J.; et al. Isolation and characterisation of KP34-a novel φKMV-like bacteriophage for Klebsiella pneumoniae. Appl. Microbiol. Biotechnol. 2011, 90, 1333–1345. [Google Scholar] [CrossRef]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Mattila, S.; Ruotsalainen, P.; Jalasvuori, M. On-demand isolation of bacteriophages against drug-resistant bacteria for personalized phage therapy. Front Microbiol. 2015, 6, 1271. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.; Zhang, Y.; Cheng, M.; Le, S.; Gu, J.; Bao, J.; Qin, J.; Guo, X.; Zhu, T. Characterization of Klebsiella pneumoniae ST11 isolates and their interactions with lytic phages. Viruses 2019, 11, 1080. [Google Scholar] [CrossRef]
- Tomita, K.; Fukai, S.; Ishitani, R.; Ueda, T.; Takeuchi, N.; Vassylyev, D.G.; Nureki, O. Structural basis for template-independent RNA polymerization. Nature 2004, 430, 700–704. [Google Scholar] [CrossRef]
- Yamashita, S.; Takeshita, D.; Tomita, K. Translocation and rotation of tRNA during template-independent RNA polymerization by tRNA nucleotidyltransferase. Structure 2014, 22, 315–325. [Google Scholar] [CrossRef]
- Kuhn, C.D.; Wilusz, J.E.; Zheng, Y.; Beal, P.A.; Joshua-Tor, L. On-enzyme refolding permits small RNA and tRNA surveillance by the CCA-adding enzyme. Cell 2015, 160, 644–658. [Google Scholar] [CrossRef]
- Augustin, M.A.; Reichert, A.S.; Betat, H.; Huber, R.; Morl, M.; Steegborn, C. Crystal structure of the human CCA-adding enzyme: Insights into template-independent polymerization. J. Mol. Biol. 2003, 328, 985–994. [Google Scholar] [CrossRef]
- Li, F.; Xiong, Y.; Wang, J.; Cho, H.D.; Tomita, K.; Weiner, A.M.; Steitz, T.A. Crystal structures of the Bacillus stearothermophilus CCA-adding enzyme and its complexes with ATP or CTP. Cell 2002, 111, 815–824. [Google Scholar] [CrossRef]
- Toh, Y.; Takeshita, D.; Numata, T.; Fukai, S.; Nureki, O.; Tomita, K. Mechanism for the definition of elongation and termination by the class II CCA-adding enzyme. EMBO J. 2009, 28, 3353–3365. [Google Scholar] [CrossRef] [PubMed]
- Toh, Y.; Takeshita, D.; Nagaike, T.; Numata, T.; Tomita, K. Mechanism for the alteration of the substrate specificities of template-independent RNA polymerases. Structure 2011, 19, 232–243. [Google Scholar] [CrossRef] [PubMed]
- De Wijn, R.; Hennig, O.; Roche, J.; Engilberge, S.; Rollet, K.; Fernandez-Millan, P.; Brillet, K.; Betat, H.; Morl, M.; Roussel, A.; et al. A simple and versatile microfluidic device for efficient biomacromolecule crystallization and structural analysis by serial crystallography. IUCrJ 2019, 6, 454–464. [Google Scholar] [CrossRef] [PubMed]
- Essoh, C.; Latino, L.; Midoux, C.; Blouin, Y.; Loukou, G.; Nguetta, S.P.; Lathro, S.; Cablanmian, A.; Kouassi, A.K.; Vergnaud, G.; et al. Investigation of a large collection of Pseudomonas aeruginosa bacteriophages collected from a single environmental source in Abidjan, Cote d’Ivoire. PLoS ONE 2015, 10, e0130548. [Google Scholar] [CrossRef] [PubMed]
- Essoh, C.; Blouin, Y.; Loukou, G.; Cablanmian, A.; Lathro, S.; Kutter, E.; Thien, H.V.; Vergnaud, G.; Pourcel, C. The susceptibility of Pseudomonas aeruginosa strains from cystic fibrosis patients to bacteriophages. PLoS ONE 2013, 8, e60575. [Google Scholar] [CrossRef]
- Oliveira, H.; Pinto, G.; Hendrix, H.; Noben, J.P.; Gawor, J.; Kropinski, A.M.; Lobocka, M.; Lavigne, R.; Azeredo, J. A lytic Providencia rettgeri virus of potential therapeutic value is a deep-branching member of the T5virus genus. Appl. Environ. Microbiol. 2017, 83, e01567-17. [Google Scholar] [CrossRef] [PubMed]
- Abelson, J.; Thomas, C.A.J. The anatomy of the T5 bacteriophage DNA molecule. J. Mol. Biol. 1966, 18, 262–291. [Google Scholar] [CrossRef]
- Rhoades, M.; Lange-Gustafson, B. Physical map of bacteriophage BF23 DNA: Terminal redundancy and localization of single-chain interruptions. J. Virol. 1979, 30, 777–786. [Google Scholar] [CrossRef]
- Glukhov, A.S.; Krutilina, A.I.; Shlyapnikov, M.G.; Severinov, K.; Lavysh, D.; Kochetkov, V.V.; McGrath, J.W.; de Leeuwe, C.; Shaburova, O.V.; Krylov, V.N.; et al. Genomic analysis of Pseudomonas putida phage tf with localized single-strand DNA interruptions. PLoS ONE 2012, 7, e51163. [Google Scholar] [CrossRef]
- Kulakov, L.A.; Ksenzenko, V.N.; Shlyapnikov, M.G.; Kochetkov, V.V.; Del Casale, A.; Allen, C.C.; Larkin, M.J.; Ceyssens, P.J.; Lavigne, R. Genomes of “phiKMV-like viruses” of Pseudomonas aeruginosa contain localized single-strand interruptions. Virology 2009, 391, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Ceyssens, P.J.; Dunon, V.; Ackermann, H.W.; Van Vaerenbergh, J.; Maes, M.; De Proft, M.; Lavigne, R. Bacteriophages LIMElight and LIMEzero of Pantoea agglomerans, belonging to the "phiKMV-like viruses". Appl. Environ. Microbiol. 2011, 77, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
- Latka, A.; Leiman, P.G.; Drulis-Kawa, Z.; Briers, Y. Modeling the architecture of depolymerase-containing receptor binding proteins in Klebsiella phages. Front Microbiol. 2019, 10, 2649. [Google Scholar] [CrossRef] [PubMed]
- Aravind, L.; Koonin, E.V. DNA polymerase beta-like nucleotidyltransferase superfamily: Identification of three new families, classification and evolutionary history. Nucleic Acids Res. 1999, 27, 1609–1618. [Google Scholar] [CrossRef]
- Kuchta, K.; Knizewski, L.; Wyrwicz, L.S.; Rychlewski, L.; Ginalski, K. Comprehensive classification of nucleotidyltransferase fold proteins: Identification of novel families and their representatives in human. Nucleic Acids Res. 2009, 37, 7701–7714. [Google Scholar] [CrossRef]
- Daniels, C.M.; Ong, S.E.; Leung, A.K. The promise of proteomics for the study of ADP-ribosylation. Mol. Cell 2015, 58, 911–924. [Google Scholar] [CrossRef]
- Holm, L.; Sander, C. DNA polymerase beta belongs to an ancient nucleotidyltransferase superfamily. Trends Biochem. Sci. 1995, 20, 345–347. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spruit, C.M.; Wicklund, A.; Wan, X.; Skurnik, M.; Pajunen, M.I. Discovery of Three Toxic Proteins of Klebsiella Phage fHe-Kpn01. Viruses 2020, 12, 544. https://doi.org/10.3390/v12050544
Spruit CM, Wicklund A, Wan X, Skurnik M, Pajunen MI. Discovery of Three Toxic Proteins of Klebsiella Phage fHe-Kpn01. Viruses. 2020; 12(5):544. https://doi.org/10.3390/v12050544
Chicago/Turabian StyleSpruit, Cindy M., Anu Wicklund, Xing Wan, Mikael Skurnik, and Maria I. Pajunen. 2020. "Discovery of Three Toxic Proteins of Klebsiella Phage fHe-Kpn01" Viruses 12, no. 5: 544. https://doi.org/10.3390/v12050544
APA StyleSpruit, C. M., Wicklund, A., Wan, X., Skurnik, M., & Pajunen, M. I. (2020). Discovery of Three Toxic Proteins of Klebsiella Phage fHe-Kpn01. Viruses, 12(5), 544. https://doi.org/10.3390/v12050544