Characterization of Molecular Cluster Detection and Evaluation of Cluster Investigation Criteria Using Machine Learning Methods and Statewide Surveillance Data in Washington State


Abstract
1. Introduction
2. Materials and Methods
2.1. Study Design and Data Collection
2.2. Variables
2.3. Analysis
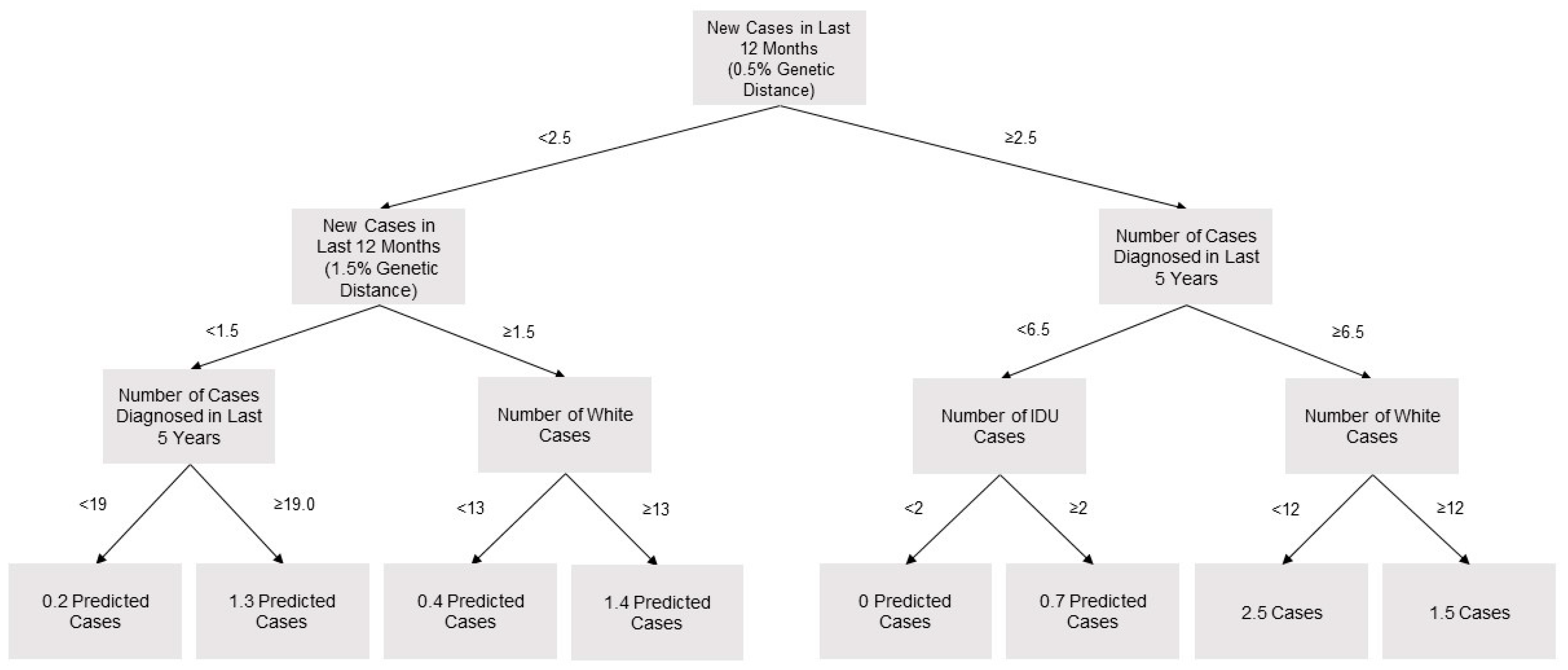
2.4. Prediction of Cluster Growth
2.5. Evaluation of Investigation Criteria
3. Results
3.1. Cluster Detection in Washington State
3.2. Predictors of Cluster Growth
3.3. Derivation and Evaluation of Investigation Criteria
4. Discussion
Author Contributions
Funding
Conflicts of Interest
References
- Centers for Disease Control and Prevention. HIV Surveillance Report, 2018 (Preliminary); 2019. Available online: https://www.cdc.gov/hiv/pdf/library/reports/surveillance/cdc-hiv-surveillance-report-2018-vol-30.pdf (accessed on 12 December 2019).
- Washington State Department of Health. Washington State HIV Surveilance Report 2019 Edition; 2019. Available online: https://www.doh.wa.gov/Portals/1/Documents/Pubs/150-030-WAHIVSurveillanceReport2019.pdf (accessed on 12 December 2019).
- Center for Disease Control and Prevention. Ending the HIV Epidemic: A Plan for America; 2019. Available online: https://www.cdc.gov/endhiv/index.html (accessed on 12 December 2019).
- Smith, D.M.; May, S.J.; Tweeten, S.; Drumright, L.; Pacold, M.E.; Kosakovsky Pond, S.L.; Pesano, R.L.; Lie, Y.S.; Richman, D.D.; Frost, S.D.W.; et al. A public health model for the molecular surveillance of HIV transmission in San Diego, California. AIDS Lond. Engl. 2009, 23, 225–232. [Google Scholar] [CrossRef] [PubMed]
- Poon, A.F.Y.; Gustafson, R.; Daly, P.; Zerr, L.; Demlow, S.E.; Wong, J.; Woods, C.K.; Hogg, R.S.; Krajden, M.; Moore, D.; et al. Near real-time monitoring of HIV transmission hotspots from routine HIV genotyping: An implementation case study. Lancet HIV 2016, 3, e231–e238. [Google Scholar] [CrossRef]
- Oster, A.M.; France, A.M.; Panneer, N.; Bañez Ocfemia, M.C.; Campbell, E.; Dasgupta, S.; Switzer, W.M.; Wertheim, J.O.; Hernandez, A.L. Identifying Clusters of Recent and Rapid HIV Transmission Through Analysis of Molecular Surveillance Data. JAIDS J. Acquir. Immune Defic. Syndr. 2018, 79, 543–550. [Google Scholar] [CrossRef] [PubMed]
- Lewis, F.; Hughes, G.J.; Rambaut, A.; Pozniak, A.; Leigh Brown, A.J. Episodic Sexual Transmission of HIV Revealed by Molecular Phylodynamics. PLoS Med. 2008, 5, e50. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. Detecting and Responding HIV Transmission Clusters: A Guide for Health Departments; 2018; p. 16. Available online: https://www.cdc.gov/hiv/pdf/funding/announcements/ps18-1802/CDC-HIV-PS18-1802-AttachmentE-Detecting-Investigating-and-Responding-to-HIV-Transmission-Clusters.pdf (accessed on 12 December 2019).
- Reuer, J.; Erly, S.; Lechtenberg, R.; Buskin, S. Issues using molecular data and the ongoing importance of HIV partner services in detecting HIV clusters – the Washington State and King County experience 2019.
- Golden, M.R.; Lechtenberg, R.; Glick, S.N.; Dombrowski, J.; Duchin, J.; Reuer, J.R.; Dhanireddy, S.; Neme, S.; Buskin, S.E. Outbreak of Human Immunodeficiency Virus Infection Among Heterosexual Persons Who Are Living Homeless and Inject Drugs — Seattle, Washington, 2018. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Wertheim, J.O.; Murrell, B.; Mehta, S.R.; Forgione, L.A.; Kosakovsky Pond, S.L.; Smith, D.M.; Torian, L.V. Growth of HIV-1 Molecular Transmission Clusters in New York City. J. Infect. Dis. 2018, 218, 1943–1953. [Google Scholar] [CrossRef] [PubMed]
- Billock, R.M.; Powers, K.A.; Pasquale, D.K.; Samoff, E.; Mobley, V.L.; Miller, W.C.; Eron, J.J.; Dennis, A.M. Prediction of HIV Transmission Cluster Growth with Statewide Surveillance Data. JAIDS J. Acquir. Immune Defic. Syndr. 2019, 80, 152–159. [Google Scholar] [CrossRef] [PubMed]
- Breiman, L. Random Forests. Mach. Learn. 2001, 45, 5–32. [Google Scholar] [CrossRef]
- Kane, M.J.; Price, N.; Scotch, M.; Rabinowitz, P. Comparison of ARIMA and Random Forest time series models for prediction of avian influenza H5N1 outbreaks. BMC Bioinformatics 2014, 15, 276. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention. CDC HIV Prevention Progress Report; Atlanta, GA, USA, 2019. Available online: https://www.cdc.gov/hiv/pdf/policies/progressreports/cdc-hiv-preventionprogressreport.pdf (accessed on 12 December 2019).
- Erly, S.; Reuer, J. Development and Impact of a New HIV Incidence Definition for Washington State; HIV/AIDS Epidemiology Report and Community Profile; 2019; pp. 24–27. Available online: https://www.kingcounty.gov/depts/health/communicable-diseases/hiv-std/patients/epidemiology/~/media/depts/health/communicable-diseases/documents/hivstd/2019-hiv-aids-epidemiology-annual-report.ashx (accessed on 12 December 2019).
- Dasgupta, S.; France, A.M.; Brandt, M.-G.; Reuer, J.; Zhang, T.; Panneer, N.; Hernandez, A.L.; Oster, A.M. Estimating Effects of HIV Sequencing Data Completeness on Transmission Network Patterns and Detection of Growing HIV Transmission Clusters. AIDS Res. Hum. Retroviruses 2019, 35, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, S.; Hall, H.I.; Hernandez, A.L.; Ocfemia, M.C.B.; Saduvala, N.; Oster, A.M. Receipt and timing of HIV drug resistance testing in six U.S. jurisdictions. AIDS Care 2017, 29, 1567–1575. [Google Scholar] [CrossRef]
- Kosakovsky Pond, S.L.; Weaver, S.; Leigh Brown, A.J.; Wertheim, J.O. HIV-TRACE (TRAnsmission Cluster Engine): A Tool for Large Scale Molecular Epidemiology of HIV-1 and Other Rapidly Evolving Pathogens. Mol. Biol. Evol. 2018, 35, 1812–1819. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Liaw, A.; Wiener, M. R News. December 2002, pp. 18–22. Available online: https://www.r-project.org/doc/Rnews/Rnews_2002-3.pdf (accessed on 12 December 2019).
- Han, S.; Yuan, B.; Liu, W. Rare Class Mining: Progress and Prospect. In Proceedings of the 2009 Chinese Conference on Pattern Recognition; IEEE: Nanjing, China, 2009; pp. 1–5. [Google Scholar]
- Cranston, K.; Alpren, C.; John, B.; Dawson, E.; Roosevelt, K.; Burrage, A.; Bryant, J.; Switzer, W.M.; Breen, C.; Peters, P.J.; et al. Notes from the Field: HIV Diagnoses Among Persons Who Inject Drugs — Northeastern Massachusetts, 2015–2018. MMWR Morb. Mortal. Wkly. Rep. 2019, 68, 253–254. [Google Scholar] [CrossRef]


{kind=link}
| Investigation Criteria | Genetic Distance Threshold | Number of Diagnoses in Past 12 Months |
|---|---|---|
| Washington State (Loose) | 1.5% | 3 |
| Washington State (Strict) | 1.5% | 5 |
| CDC (Strict) | 0.5% | 5 |
| Variable | 2015 | 2016 | 2017 | 2018 | Total |
|---|---|---|---|---|---|
| Number of Analyzed Clusters a | 95 | 95 | 103 | 107 | 107 |
| Number of New Cases WA State | 459 | 436 | 441 | 511 | 1847 |
| New Cases with Analyzable Genotype b | 276 (60%) | 238 (55%) | 252 (57%) | 292 (57%) | 1058 (57%) |
| Number of New Cases in All Clusters | 156 (34%) | 112 (26%) | 134 (30%) | 148 (29%) | 550 (29%) |
| Number of New Cases in Analyzed Clusters | 85 (19%) | 84 (19%) | 112 (25%) | 120 (23%) | 401 (22%) |
| Cumulative Prevalent Cases in WA State c | 12,631 | 13,051 | 13,424 | 13,764 | 15,150 |
| Prevalent Cases with Analyzable Genotype b | 6320 (50%) | 6458 (49%) | 6583 (49%) | 6733 (49%) | 7373 (49%) |
| Number of Prevalent Cases in All Clusters | 2238 (18%) | 2310 (18%) | 2389 (18%) | 2476 (18%) | 2680 (18%) |
| Number of Prevalent Cases in Analyzed Clusters | 824 (7%) | 897 (7%) | 983 (7%) | 1078 (8%) | 1157 (8%) |
| Median Days from Dx to Specimen Collection | 17 (6–30) | 14 (7–31) | 15 (7–31) | 11 (4–33) | 14 (6–31) |
| Median Days from Dx to TRACE Analysis | 821 (734–921) | 468 (353–576) | 201 (143–255) | 109 (77–137) | 291 (138–714) |
| Variable | Value | All Cases | Cases with PR or RT Sequence | p-Value b | Study Population a | p-Value b |
|---|---|---|---|---|---|---|
| N | 15,150 | 7499 | - | 1157 | - | |
| Time Since Diagnosis | <1 Year | 540 (4%) | 297 (4%) | <0.01 | 121 (11%) | <0.01 |
| 1–5 Years | 2557 (17%) | 1393 (19%) | 417 (36%) | |||
| 6–10 Years | 2870 (19%) | 1797 (24%) | 433 (37%) | |||
| >10 Years | 7846 (51%) | 3886 (53%) | 186 (16%) | |||
| Gender | Female | 2278 (15%) | 1108 (15%) | <0.01 | 73 (6%) | <0.01 |
| Transgender Male | 14 (0%) | 7 (0%) | 0 (0%) | |||
| Male | 12,727 (84%) | 6173 (83%) | 1072 (93%) | |||
| Transgender Female | 131 (1%) | 85 (1%) | 12 (1%) | |||
| Race | WHITE | 8747 (58%) | 4046 (55%) | <0.01 | 742 (64%) | <0.01 |
| BLACK | 2588 (17%) | 1273 (17%) | 99 (9%) | |||
| HISP | 2152 (14%) | 1118 (15%) | 194 (17%) | |||
| ASIAN | 516 (3%) | 248 (3%) | 31 (3%) | |||
| HAW/PI | 69 (0%) | 43 (1%) | 10 (1%) | |||
| AI/AN | 156 (1%) | 81 (1%) | 12 (1%) | |||
| MULTI | 915 (6%) | 564 (8%) | 69 (6%) | |||
| UNK | 7 (0%) | 0 (0%) | 0 (0%) | |||
| Age (31 December, 2018) | <13 | 40 (0%) | 12 (0%) | <0.01 | 0 (0%) | <0.01 |
| 13–24 | 328 (2%) | 171 (2%) | 56 (5%) | |||
| 25–34 | 2143 (14%) | 1143 (16%) | 385 (33%) | |||
| 35–44 | 3089 (20%) | 1680 (23%) | 340 (29%) | |||
| 45–54 | 4456 (29%) | 2219 (30%) | 250 (22%) | |||
| 55–64 | 3728 (25%) | 1677 (23%) | 108 (9%) | |||
| >64 | 1366 (9%) | 471 (6%) | 18 (2%) | |||
| Risk | MSM | 9234 (61%) | 4392 (60%) | <0.01 | 854 (74%) | <0.01 |
| IDU | 924 (6%) | 502 (7%) | 79 (7%) | |||
| MSM/IDU | 1465 (10%) | 813 (11%) | 137 (12%) | |||
| TRANFUS | 21 (0%) | 9 (0%) | 0 (0%) | |||
| HEMO | 21 (0%) | 6 (0%) | 0 (0%) | |||
| HETERO | 1787 (12%) | 875 (12%) | 38 (3%) | |||
| PED | 140 (1%) | 73 (1%) | 0 (0%) | |||
| NIR | 1549 (11%) | 703 (10%) | (4%) |
| Variable | Value | Cluster-Months | Absolute 3 Month Cluster Growth, Mean (5% CI) a | p-Value | Cluster Growth Per 100 Person-Months, Mean (95% CI) a | p-Value |
|---|---|---|---|---|---|---|
| Total Population | All | 2318 | 0.24 (0.18–0.33) | 1.22 (0.86–1.73) | ||
| Viremic Individuals | 0 | 344 | 0.13 (0.08–0.20) | 0.023 | 1.20 (0.72–2.01) | 0.212 |
| 1 | 533 | 0.22 (0.15–0.33) | 1.69 (1.04–2.76) | |||
| 2 | 462 | 0.18 (0.12–0.27) | 1.00 (0.65–1.55) | |||
| 3+ | 530 | 0.38 (0.26–0.57) | 0.96 (0.55–1.68) | |||
| Percent Viremic (Quartiles) | <12% | 353 | 0.12 (0.08–0.20) | 0.024 | 1.17(0.70–1.96) | 0.248 |
| 12–25% | 692 | 0.32 (0.23–0.46) | 0.92 (0.63–1.35) | |||
| 26–35% | 496 | 0.23 (0.15–0.35) | 1.19 (0.76–1.88) | |||
| ≥35% | 328 | 0.21 (0.13–0.36) | 1.97 (1.05–3.69) | |||
| Cluster Size (Quartiles) | ≤3 | 517 | 0.17 (0.11–0.27) | 0.010 | 2.07 (1.29–3.34) | 0.011 |
| 4–5 | 430 | 0.13 (0.08–0.20) | 0.93 (0.60–1.45) | |||
| 6–12 | 498 | 0.30 (0.19–0.47) | 1.26 (0.80–2.00) | |||
| >12 | 424 | 0.37 (0.24–0.58) | 0.43 (0.33–0.58) | |||
| % White | <16% | 939 | 0.23 (0.15–0.35) | 0.685 | 1.18 (0.71–1.95) | 0.774 |
| ≥18% | 930 | 0.25 (0.18–0.35) | 1.27 (0.88–1.85) | |||
| % Female | 0% | 1343 | 0.21 (0.15–0.31) | 0.203 | 1.18 (0.76–1.82) | 0.727 |
| >0% | 526 | 0.31 (0.21–0.47) | 1.34 (0.76–2.35) | |||
| % IDU | <16% | 943 | 0.24 (0.17–0.35) | 0.966 | 0.93 (0.70–1.23) | 0.165 |
| ≥15% | 926 | 0.24 (0.15–0.38) | 1.52 (0.90–2.58) | |||
| % Late | <13% | 932 | 0.24 (0.16–0.35) | 0.933 | 1.08 (0.73–1.62) | 0.522 |
| ≥13% | 937 | 0.24 (0.16–0.38) | 1.36 (0.80–2.33) | |||
| % Diagnosed in Past 5 Years | <25% | 363 | 0.19 (0.13–0.29) | 0.721 | 1.05 (0.56–1.98) | 0.572 |
| 25–50% | 702 | 0.26 (0.17–0.40) | 1.09 (0.67–1.79) | |||
| 51–66% | 349 | 0.26 (0.17–0.38) | 1.45 (0.95–2.21) | |||
| >66% | 455 | 0.24 (0.13–0.46) | 1.39 (0.85–2.25) | |||
| 3 Cases in Previous 12 Months (0.015) | No | 1685 | 0.18 (0.15–0.22) | 0.022 | 1.09 (0.83–1.41) | 0.253 |
| Yes | 184 | 0.78 (0.47–1.27) | 2.49 (0.98–6.33) | |||
| 5 Cases in Previous 12 Months (0.015) | No | 1819 | 0.21 (0.17–0.27) | 0.088 | 1.16 (0.86–1.56) | 0.263 |
| Yes | 50 | 1.26 (0.89–1.78) | 3.69 (1.49–9.14) | |||
| 3 Cases in Previous 12 Months (0.005) | No | 1818 | 0.21 (0.17–0.27) | 0.058 | 1.17 (0.85–1.62) | 0.224 |
| Yes | 51 | 1.29 (0.74–2.26) | 3.00 (1.19–7.58) | |||
| 5 Cases in Previous 12 Months (0.005) | No | 1847 | 0.23 (0.17–0.29) | 0.116 | 1.20 (0.86–1.68) | 0.388 |
| Yes | 22 | 1.50 (0.79–2.86) | 2.93 (0.79–10.86) |
| Variable | Value | Cluster-Months | Absolute 12-Month Cluster Growth, Mean (95% CI) a | p-value | Cluster Growth Per 100 Person-Months, Mean (95% CI) a | p-Value |
|---|---|---|---|---|---|---|
| Total Population | All | 2318 | 1.02 (0.75–1.38) | 1.27 (0.89–1.81) | ||
| Viremic Individuals | 0 | 344 | 0.62 (0.31–1.25) | 0.081 | 1.35 (0.60–3.05) | 0.369 |
| 1 | 533 | 0.85 (0.59–1.23) | 1.61 (1.07–2.42) | |||
| 2 | 462 | 0.82 (0.57–1.20) | 1.18 (0.78–1.81) | |||
| 3+ | 530 | 1.62 (1.09–2.41) | 0.95 (0.56–1.62) | |||
| Percent Viremic (Quartiles) | <12% | 353 | 0.65 (0.34–1.25) | 0.216 | 1.33 (0.59–2.98) | 0.056 |
| 12%–25% | 692 | 1.28 (0.87–1.87) | 0.83 (0.56–1.22) | |||
| 26%–35% | 496 | 0.95 (0.65–1.39) | 1.40 (0.95–2.07) | |||
| ≥35% | 328 | 0.98 (0.55–1.77) | 1.94 (1.06–3.53) | |||
| Cluster Size (Quartiles) | ≤3 | 517 | 0.74 (0.48–1.17) | 0.029 | 2.14 (1.37–3.34) | 0.014 |
| 4–5 | 430 | 0.53 (0.33–0.84) | 0.94 (0.60–1.49) | |||
| 6–12 | 498 | 1.28 (0.79–2.09) | 1.37 (0.81–2.32) | |||
| >12 | 424 | 1.55 (0.96–2.50) | 0.42 (0.32–0.55) | |||
| % White | <16% | 939 | 0.87 (0.58–1.20) | 0.237 | 1.15 (0.71–1.84] | 0.521 |
| ≥18% | 930 | 1.18 (0.80–1.72) | 1.40 (0.89–2.20) | |||
| % Female | 0% | 1343 | 0.83 (0.57–1.22) | 0.072 | 1.04 (0.69–1.55) | 0.184 |
| >0% | 526 | 1.50 (0.98–2.30) | 1.87 (1.02–3.41) | |||
| % IDU | <16% | 943 | 0.99 (0.66–1.48) | 0.836 | 0.84 (0.63–1.11) | 0.050 |
| ≥15% | 926 | 1.05 (0.68–1.62) | 1.71 (1.05–2.78) | |||
| % Late | <13% | 932 | 1.03 (0.68–1.57) | 0.940 | 1.11 (0.66–1.86) | 0.472 |
| ≥13% | 937 | 1.01 (0.66–1.55) | 1.43 (0.89–2.31) | |||
| % Diagnosed in Past 5 Years | <25% | 363 | 0.87 (0.54–1.39) | 0.670 | 1.28 (0.55–2.97) | 0.143 |
| 25–50% | 702 | 1.02 (0.63–1.65) | 0.94 (0.61–1.45) | |||
| 51–66% | 349 | 1.27 (0.83–1.96) | 1.96 (1.22–3.15) | |||
| >66% | 455 | 0.95 (0.49–1.87) | 1.24 (0.70–2.19) | |||
| 3 Cases in Previous 12 Months (0.015) | No | 1685 | 0.82 (0.64–1.05) | 0.029 | 1.15 (0.85–1.58) | 0.252 |
| Yes | 184 | 2.88 (1.77–4.69) | 2.33 (1.01–5.42) | |||
| 5 Cases in Previous 12 Months (0.015) | No | 1819 | 0.91 (0.7–1.19) | 0.105 | 1.20 (0.86–1.67) | 0.337 |
| Yes | 50 | 4.98 [3.28–7.55) | 3.75 (1.32–10.67) | |||
| 3 Cases in Previous 12 Months (0.005) | No | 1818 | 0.91 (0.7–1.18) | 0.041 | 1.20 (0.86–1.68) | 0.154 |
| Yes | 51 | 5.12 [3.4–7.69) | 3.74 (1.75–8.00) | |||
| 5 Cases in Previous 12 Months (0.005) | No | 1847 | 0.97 (0.73–1.29) | 0.098 | 1.26 (0.89–1.79) | 0.507 |
| Yes | 22 | 4.91 (2.7–8.92) | 2.11 (0.62–7.19) |
| Cluster Investigation Criteria | Number of Clusters Reaching Investigation Criteria | Prevalent Cases at First Indicated Investigation, Mean (95% CI) | Absolute 3 Month Cluster Growth at First Indicated Investigation, Mean (95% CI) a |
| 3 Cases at 0.015 | 17 | 20.5 (8.3–32.6) | 1.4 (0.8–2.0) |
| 5 Cases at 0.015 | 9 | 28.0 (4.9–50.2) | 1.4 (0.5–2.3) |
| 3 Cases at 0.005 | 10 | 24.6 (6.6–42.6) | 1.6 (0.8–2.4) |
| 5 Cases at 0.005 | 6 | 28.3 (−5.7–62.4) | 2.0 (0.5–3.4) |
| >0.9 Predicted Cases in 3 Months (Random Forest) b | 14 | 19.9 (16.8–23.1) | 1.3 (1.1–1.4) |
| >2.3 Predicted Cases in 3 Months (Random Forest) b | 4 | 31.8 (20.5–42.9) | 2.3 (1.3–3.2) |
| Cluster Investigation Criteria | Number of Clusters Reaching Investigation Criteria | Prevalent Cases at First Indicated Investigation, Mean (95% CI) | Cluster Growth Per 100 Person-Months at First Indicated Investigation, Mean (95% CI) a |
| 3 Cases at 0.015 | 17 | 20.5 (8.3–32.6) | 1.5 (1.3–7.7) |
| 5 Cases at 0.015 | 9 | 28.0 (4.9–50.2) | 2.6 (0.7–4.4) |
| 3 Cases at 0.005 | 10 | 24.6 (3.8–45.4) | 5.3 (−0.3–10.9) |
| 5 Cases at 0.005 | 6 | 28.3 (−5.7–62.4) | 3.1 (0.8–5.4) |
| >0.14 Predicted Cases Per 100 Person-Months (Random Forest) b | 15 | 7.5 (6.1–8.8) | 3.4 (2.8–4.0) |
| >0.42 Predicted Cases Per 100 Person-Months (Random Forest) b | 1 | 2 (NA) | 0 (NA) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Erly, S.J.; Herbeck, J.T.; Kerani, R.P.; Reuer, J.R. Characterization of Molecular Cluster Detection and Evaluation of Cluster Investigation Criteria Using Machine Learning Methods and Statewide Surveillance Data in Washington State. Viruses 2020, 12, 142. https://doi.org/10.3390/v12020142
Erly SJ, Herbeck JT, Kerani RP, Reuer JR. Characterization of Molecular Cluster Detection and Evaluation of Cluster Investigation Criteria Using Machine Learning Methods and Statewide Surveillance Data in Washington State. Viruses. 2020; 12(2):142. https://doi.org/10.3390/v12020142
Chicago/Turabian StyleErly, Steven J., Joshua T. Herbeck, Roxanne P. Kerani, and Jennifer R. Reuer. 2020. "Characterization of Molecular Cluster Detection and Evaluation of Cluster Investigation Criteria Using Machine Learning Methods and Statewide Surveillance Data in Washington State" Viruses 12, no. 2: 142. https://doi.org/10.3390/v12020142
APA StyleErly, S. J., Herbeck, J. T., Kerani, R. P., & Reuer, J. R. (2020). Characterization of Molecular Cluster Detection and Evaluation of Cluster Investigation Criteria Using Machine Learning Methods and Statewide Surveillance Data in Washington State. Viruses, 12(2), 142. https://doi.org/10.3390/v12020142