
Autochthonous Transmission of West Nile Virus by a New Vector in Iran, Vector-Host Interaction Modeling and Virulence Gene Determinants



 and
and
Abstract
1. Introduction
2. Material and Methods
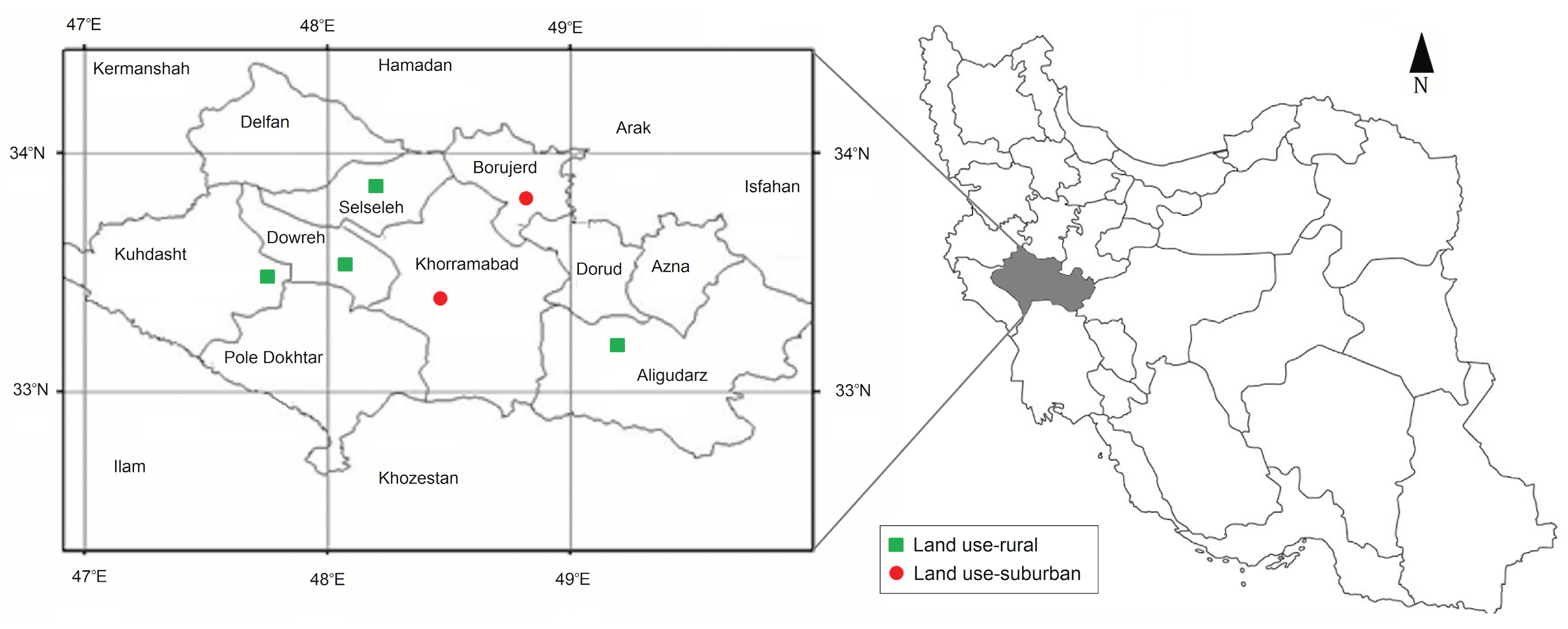
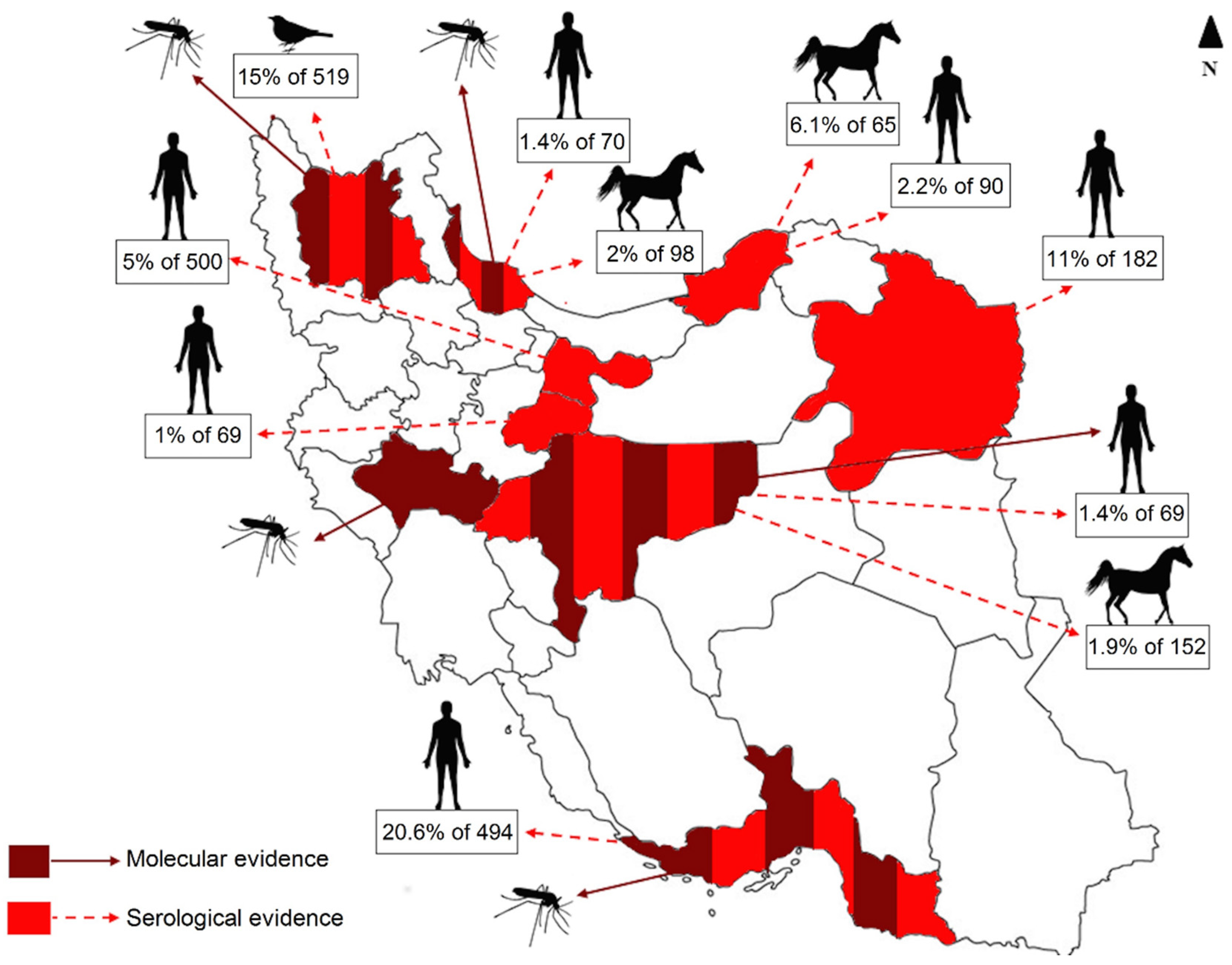
2.1. Study Area and Sampling
2.2. Morphological and Molecular Identification of Mosquitoes
2.3. Blood Source Identification
2.4. Panel PCR for Arbovirus Screening
2.5. Polyprotein Characterization
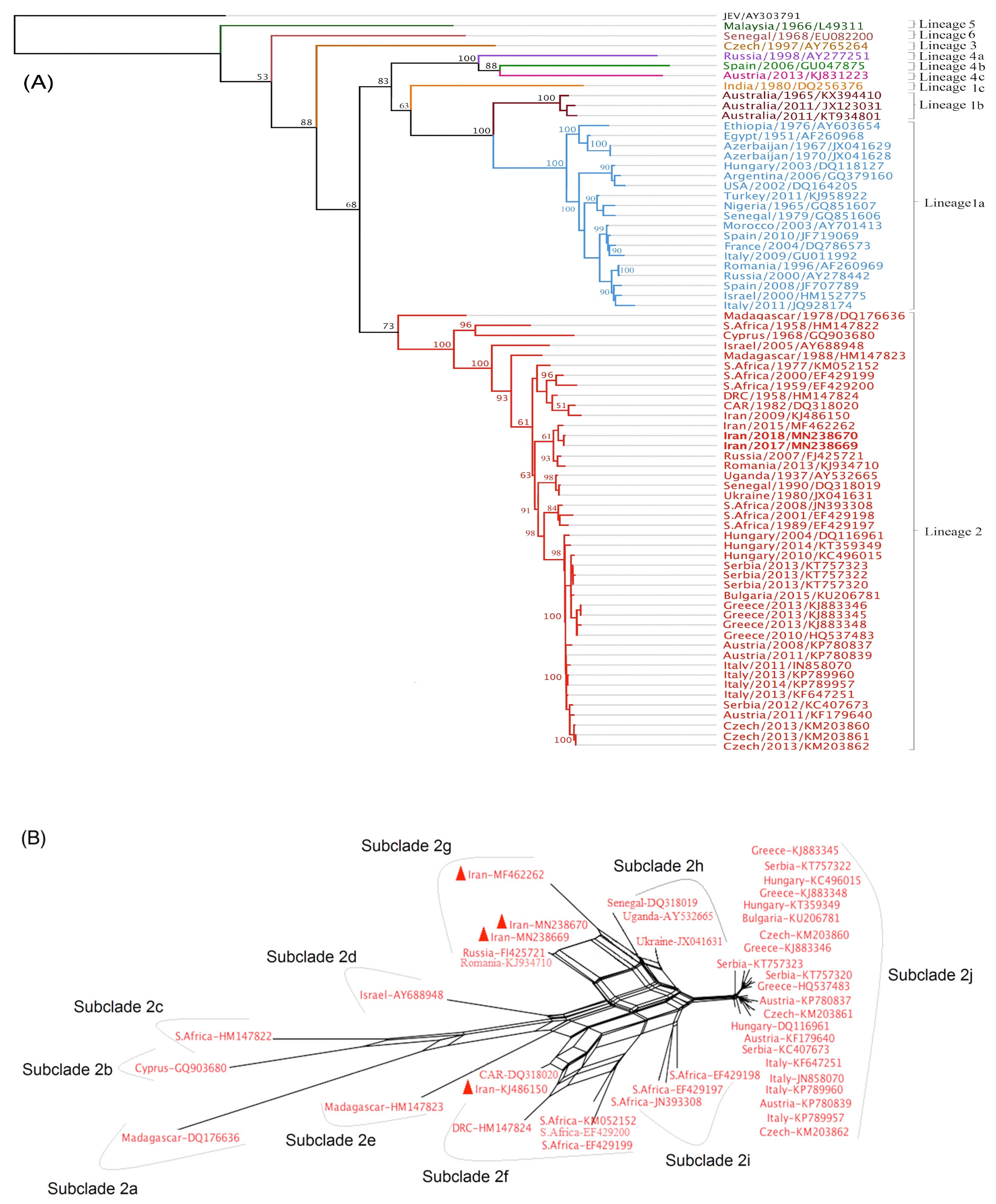
2.6. Full Genome-Based Phylogenetic Tree Construction
3. Results
3.1. Mosquito Population Dynamic
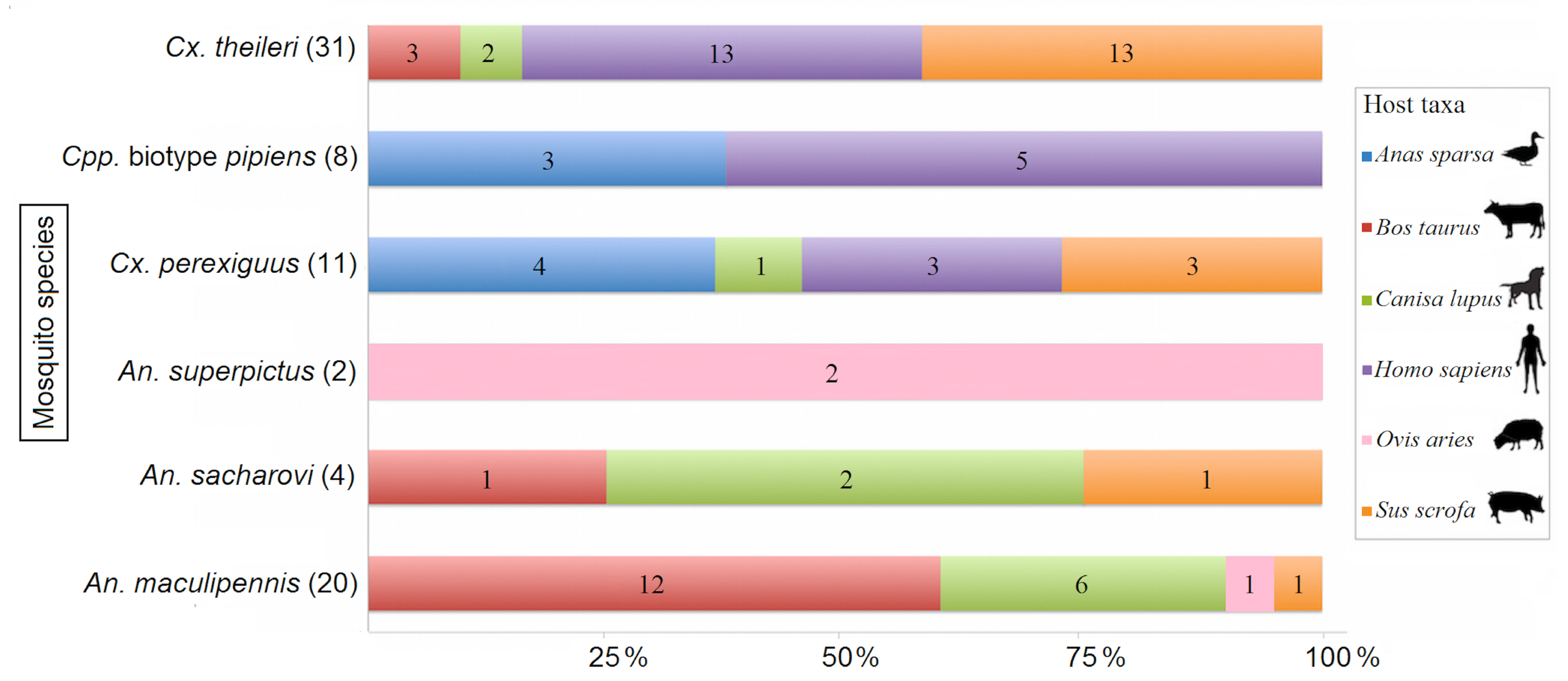
3.2. Host-Vector Interaction
3.3. WNV Prevalence in Mosquito Specimens
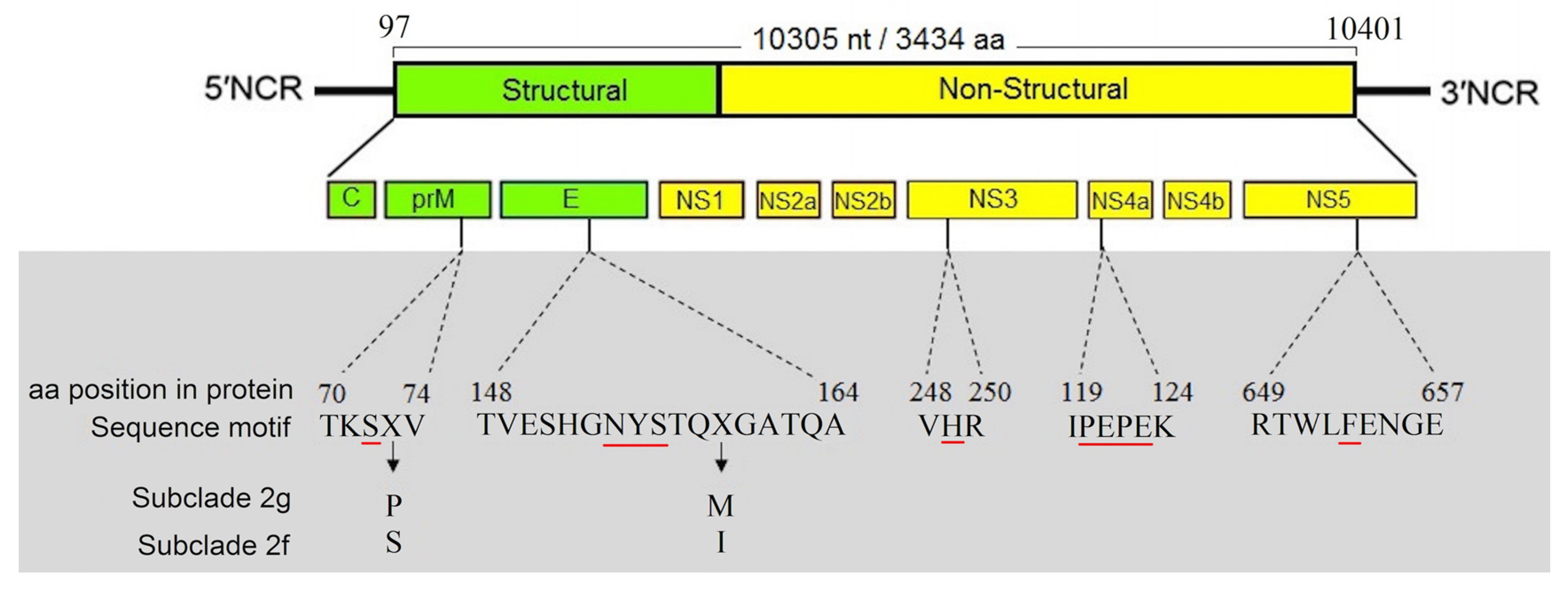
3.4. Characterization of Full-Length Genome and Amino Acid Motifs
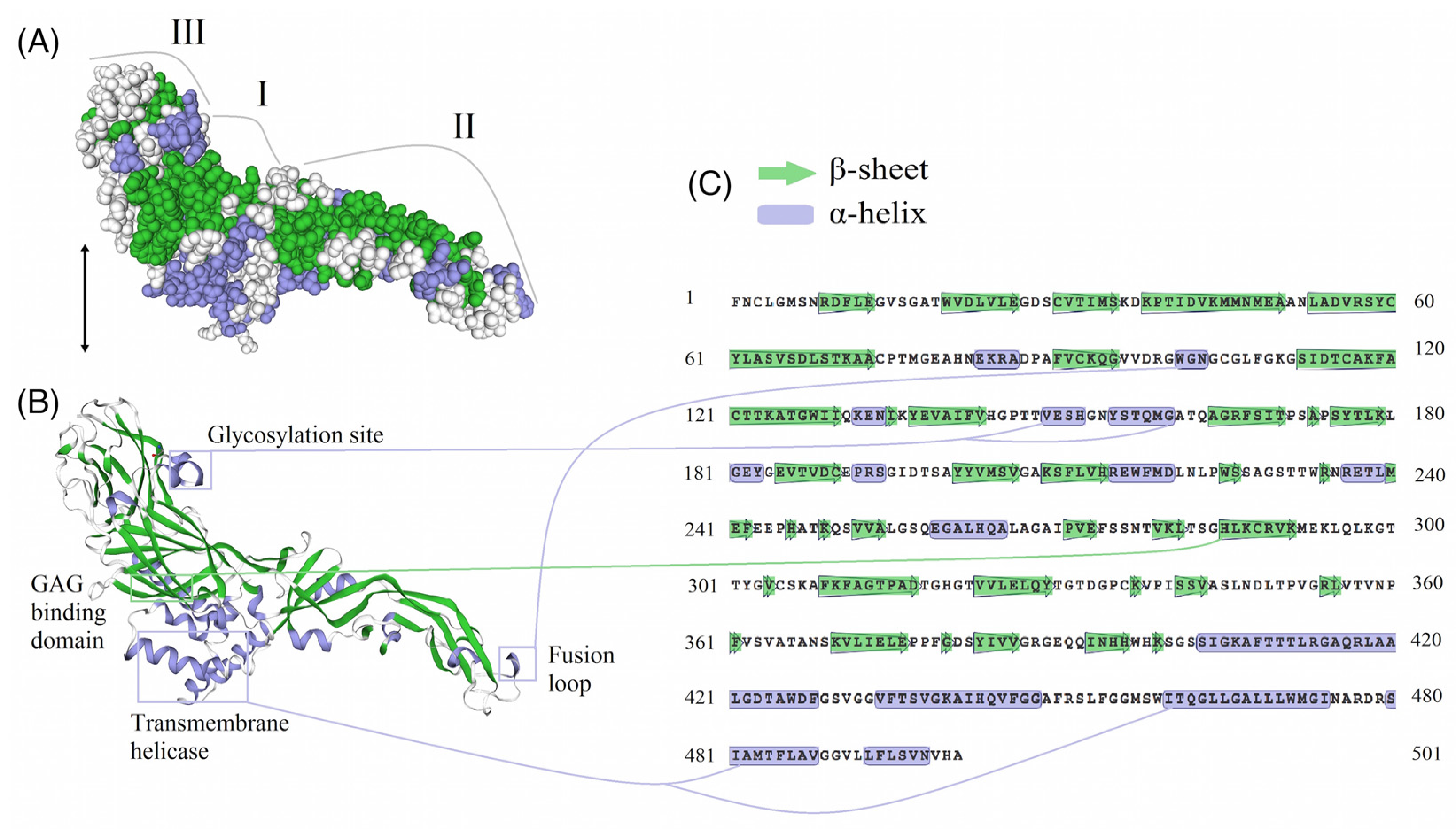
3.5. E-Protein Molecular Architecture
3.6. Genetic Determinants of Virulence in Iranian WNV Strains
3.7. Evolutionary Tree of WNV Based on Complete Nucleotide Sequences
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kayedi, M.H.; Chinikar, S.; Mostafavi, E.; Khakifirouz, S.; Jalali, T.; Hosseini-Chegeni, A.; Naghizadeh, A.; Niedrig, M.; Fooks, A.R.; Shahhosseini, N. Crimean–congo hemorrhagic fever virus clade iv (asia 1) in ticks of western iran. J. Med. Entomol. 2015, 52, 1144–1149. [Google Scholar] [CrossRef]
- Hubálek, Z.; Rudolf, I.; Nowotny, N. Arboviruses pathogenic for domestic and wild animals. In Advances in Virus Research; Elsevier: Amsterdam, The Netherlands, 2014; Volume 89, pp. 201–275. [Google Scholar]
- Chinikar, S.; Ghiasi, S.M.; Shah-Hosseini, N.; Mostafavi, E.; Moradi, M.; Khakifirouz, S.; Varai, F.S.R.; Rafigh, M.; Jalali, T.; Goya, M.M. Preliminary study of dengue virus infection in iran. Travel Med. Infect. Dis. 2013, 11, 166–169. [Google Scholar] [CrossRef]
- Chinikar, S.; Shah-Hosseini, N.; Mostafavi, E.; Moradi, M.; Khakifirouz, S.; Jalali, T.; Goya, M.M.; Shirzadi, M.R.; Zainali, M.; Fooks, A.R. Seroprevalence of west nile virus in iran. Vector Borne Zoonotic Dis. 2013, 13, 586–589. [Google Scholar] [CrossRef]
- Weissenböck, H.; Kolodziejek, J.; Url, A.; Lussy, H.; Rebel-Bauder, B.; Nowotny, N. Emergence of usutu virus, an african mosquito-borne flavivirus of the japanese encephalitis virus group, central europe. Emerg. Infect. Dis. 2002, 8, 652. [Google Scholar] [CrossRef] [PubMed]
- Bakonyi, T.; Ivanics, É.; Erdélyi, K.; Ursu, K.; Ferenczi, E.; Weissenböck, H.; Nowotny, N. Lineage 1 and 2 strains of encephalitic west nile virus, central europe. Emerg. Infect. Dis. 2006, 12, 618. [Google Scholar] [CrossRef] [PubMed]
- Shahhosseini, N.; Chinikar, S. Genetic evidence for circulation of kunjin-related west nile virus strain in iran. J. Vector Borne Dis. 2016, 53, 384. [Google Scholar] [PubMed]
- Shahhosseini, N.; Chinikar, S.; Moosa-Kazemi, S.H.; Sedaghat, M.M.; Kayedi, M.H.; Lühken, R.; Schmidt-Chanasit, J. West nile virus lineage—2 in culex specimens from iran. Trop. Med. Int. Health 2017, 22, 1343–1349. [Google Scholar] [CrossRef]
- Chinikar, S.; Javadi, A.; Ataei, B.; Shakeri, H.; Moradi, M.; Mostafavi, E.; Ghiasi, S.M. Detection of west nile virus genome and specific antibodies in iranian encephalitis patients. Epidemiol. Infect. 2012, 140, 1525–1529. [Google Scholar] [CrossRef]
- Nash, D.; Mostashari, F.; Fine, A.; Miller, J.; O’Leary, D.; Murray, K.; Huang, A.; Rosenberg, A.; Greenberg, A.; Sherman, M. The outbreak of west nile virus infection in the new york city area in 1999. N. Engl. J. Med. 2001, 344, 1807–1814. [Google Scholar] [CrossRef]
- Domanović, D.; Gossner, C.M.; Lieshout-Krikke, R.; Mayr, W.; Baroti-Toth, K.; Dobrota, A.M.; Escoval, M.A.; Henseler, O.; Jungbauer, C.; Liumbruno, G. West nile and usutu virus infections and challenges to blood safety in the european union. Emerg. Infect. Dis. 2019, 25, 1050. [Google Scholar] [CrossRef]
- Eidson, M.; Kramer, L.; Stone, W.; Hagiwara, Y.; Schmit, K.; Team, N.Y.S.W.N.V.A.S. Dead bird surveillance as an early warning system for west nile virus. Emerg. Infect. Dis. 2001, 7, 631. [Google Scholar] [CrossRef] [PubMed]
- Hughes, J.M.; Wilson, M.E.; Sejvar, J.J. The long-term outcomes of human west nile virus infection. Clin. Infect. Dis. 2007, 44, 1617–1624. [Google Scholar] [CrossRef]
- Lanciotti, R.; Roehrig, J.; Deubel, V.; Smith, J.; Parker, M.; Steele, K.; Crise, B.; Volpe, K.; Crabtree, M.; Scherret, J. Origin of the west nile virus responsible for an outbreak of encephalitis in the northeastern united states. Science 1999, 286, 2333–2337. [Google Scholar] [CrossRef] [PubMed]
- Rizzoli, A.; Jiménez-Clavero, M.A.; Barzon, L.; Cordioli, P.; Figuerola, J.; Koraka, P.; Martina, B.; Moreno, A.; Nowotny, N.; Pardigon, N. The challenge of west nile virus in europe: Knowledge gaps and research priorities. EuroSurveill 2015, 20, 28–42. [Google Scholar] [CrossRef] [PubMed]
- Bondre, V.P.; Jadi, R.; Mishra, A.; Yergolkar, P.; Arankalle, V. West nile virus isolates from india: Evidence for a distinct genetic lineage. J. Gen. Virol. 2007, 88, 875–884. [Google Scholar] [CrossRef] [PubMed]
- Bakonyi, T.; Ferenczi, E.; Erdélyi, K.; Kutasi, O.; Csörgő, T.; Seidel, B.; Weissenböck, H.; Brugger, K.; Bán, E.; Nowotny, N. Explosive spread of a neuroinvasive lineage 2 west nile virus in central europe, 2008/2009. Vet. Microbiol. 2013, 165, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Bakonyi, T.; Hubálek, Z.; Rudolf, I.; Nowotny, N. Novel flavivirus or new lineage of west nile virus, central europe. Emerg. Infect. Dis. 2005, 11, 225. [Google Scholar] [CrossRef]
- Lvov, D.; Butenko, A.; Gromashevsky, V.; Kovtunov, A.; Prilipov, A.; Kinney, R.; Aristova, V.; Dzharkenov, A.; Samokhvalov, E.; Savage, H. West nile virus and other zoonotic viruses in russia: Examples of emerging-reemerging situations. In Emergence and Control of Zoonotic Viral Encephalitides; Springer: Berlin/Heidelberg, Germany, 2004; pp. 85–96. [Google Scholar]
- Vázquez, A.; Sánchez-Seco, M.P.; Ruiz, S.; Molero, F.; Hernández, L.; Moreno, J.; Magallanes, A.; Tejedor, C.G.; Tenorio, A. Putative new lineage of west nile virus, spain. Emerg. Infect. Dis. 2010, 16, 549. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.; Williams, D. The zoonotic flaviviruses of southern, south—Eastern and eastern asia, and australasia: The potential for emergent viruses. Zoonoses Public Health 2009, 56, 338–356. [Google Scholar] [CrossRef]
- Fall, G.; Diallo, M.; Loucoubar, C.; Faye, O. Vector competence of culex neavei and culex quinquefasciatus (diptera: Culicidae) from senegal for lineages 1, 2, koutango and a putative new lineage of west nile virus. Am. J. Trop. Med. Hyg. 2014, 90, 747–754. [Google Scholar] [CrossRef]
- Pachler, K.; Lebl, K.; Berer, D.; Rudolf, I.; Hubalek, Z.; Nowotny, N. Putative new west nile virus lineage in uranotaenia unguiculata mosquitoes, austria, 2013. Emerg. Infect. Dis. 2014, 20, 2119. [Google Scholar] [CrossRef] [PubMed]
- Beasley, D.W.; Li, L.; Suderman, M.T.; Barrett, A.D. Mouse neuroinvasive phenotype of west nile virus strains varies depending upon virus genotype. Virology 2002, 296, 17–23. [Google Scholar] [CrossRef]
- Beasley, D.W.; Whiteman, M.C.; Zhang, S.; Huang, C.Y.-H.; Schneider, B.S.; Smith, D.R.; Gromowski, G.D.; Higgs, S.; Kinney, R.M.; Barrett, A.D. Envelope protein glycosylation status influences mouse neuroinvasion phenotype of genetic lineage 1 west nile virus strains. J. Virol. 2005, 79, 8339–8347. [Google Scholar] [CrossRef] [PubMed]
- Setoh, Y.; Prow, N.; Hobson-Peters, J.; Lobigs, M.; Young, P.; Khromykh, A.; Hall, R. Identification of residues in west nile virus pre-membrane protein that influence viral particle secretion and virulence. J. Gen. Virol. 2012, 93, 1965–1975. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Jia, R.; Shen, H.; Wang, M.; Yin, Z.; Cheng, A. Structures and functions of the envelope glycoprotein in flavivirus infections. Viruses 2017, 9, 338. [Google Scholar] [CrossRef]
- Kozlovskaya, L.; Osolodkin, D.; Shevtsova, A.; Romanova, L.I.; Rogova, Y.; Dzhivanian, T.; Lyapustin, V.; Pivanova, G.; Gmyl, A.; Palyulin, V. Gag-binding variants of tick-borne encephalitis virus. Virology 2010, 398, 262–272. [Google Scholar] [CrossRef]
- Lee, E.; Hall, R.A.; Lobigs, M. Common e protein determinants for attenuation of glycosaminoglycan-binding variants of japanese encephalitis and west nile viruses. J. Virol. 2004, 78, 8271–8280. [Google Scholar] [CrossRef]
- Brault, A.C.; Huang, C.Y.; Langevin, S.A.; Kinney, R.M.; Bowen, R.A.; Ramey, W.N.; Panella, N.A.; Holmes, E.C.; Powers, A.M.; Miller, B.R. A single positively selected west nile viral mutation confers increased virogenesis in american crows. Nat. Genet. 2007, 39, 1162–1166. [Google Scholar] [CrossRef]
- Fall, G.; Di Paola, N.; Faye, M.; Dia, M.; de Melo Freire, C.C.; Loucoubar, C.; de Andrade Zanotto, P.M.; Faye, O. Biological and phylogenetic characteristics of west african lineages of west nile virus. PLoS Negl. Trop. Dis. 2017, 11, e0006078. [Google Scholar] [CrossRef]
- Van Slyke, G.A.; Ciota, A.T.; Willsey, G.G.; Jaeger, J.; Shi, P.-Y.; Kramer, L.D. Point mutations in the west nile virus (flaviviridae; flavivirus) rna-dependent rna polymerase alter viral fitness in a host-dependent manner in vitro and in vivo. Virology 2012, 427, 18–24. [Google Scholar] [CrossRef]
- Shahhosseini, N.; Wong, G.; Frederick, C.; Kobinger, G.P. Mosquito species composition and abundance in quebec, eastern Canada. J. Med. Entomol. 2020, 57, 1025–1031. [Google Scholar] [CrossRef] [PubMed]
- Azari-Hamidian, S.; Harbach, R.E. Keys to the adult females and fourth-instar larvae of the mosquitoes of Iran (diptera: Culicidae). Zootaxa 2009, 2078, 1–33. [Google Scholar] [CrossRef]
- Shahhosseini, N.; Frederick, C.; Racine, T.; Kobinger, G.P.; Wong, G. Modeling host-feeding preference and molecular systematics of mosquitoes in different ecological niches in Canada. Acta Trop. 2020, 105734. [Google Scholar] [CrossRef]
- Shahhosseini, N.; Kayedi, M.H.; Sedaghat, M.M.; Racine, T.; Kobinger, G.P.; Moosa-Kazemi, S.H. DNA barcodes corroborating identification of mosquito species and multiplex real-time pcr differentiating culex pipiens complex and culex torrentium in Iran. PLoS ONE 2018, 13, e0207308. [Google Scholar] [CrossRef] [PubMed]
- Börstler, J.; Jöst, H.; Garms, R.; Krüger, A.; Tannich, E.; Becker, N.; Schmidt-Chanasit, J.; Lühken, R. Host-feeding patterns of mosquito species in germany. Parasit. Vectors 2016, 9, 318. [Google Scholar] [CrossRef] [PubMed]
- Shahhosseini, N.; Friedrich, J.; Moosa-Kazemi, S.H.; Sedaghat, M.M.; Kayedi, M.H.; Tannich, E.; Schmidt-Chanasit, J.; Lühken, R. Host-feeding patterns of culex mosquitoes in Iran. Parasit. Vectors 2018, 11, 669. [Google Scholar] [CrossRef] [PubMed]
- Scheuch, D.E.; Schäfer, M.; Eiden, M.; Heym, E.C.; Ziegler, U.; Walther, D.; Schmidt-Chanasit, J.; Keller, M.; Groschup, M.H.; Kampen, H. Detection of usutu, sindbis, and batai viruses in mosquitoes (diptera: Culicidae) collected in germany, 2011–2016. Viruses 2018, 10, 389. [Google Scholar] [CrossRef]
- Charrel, R.N.; Bichaud, L.; de Lamballerie, X. Emergence of toscana virus in the mediterranean area. World J. Virol. 2012, 1, 135. [Google Scholar] [CrossRef]
- Bourhy, H.; Cowley, J.; Larrous, F.; Holmes, E.; Walker, P. Phylogenetic relationships among rhabdoviruses inferred using the l polymerase gene. J. Gen. Virol. 2005, 86, 2849–2858. [Google Scholar] [CrossRef]
- Shah-Hosseini, N.; Chinikar, S.; Ataei, B.; Fooks, A.R.; Groschup, M.H. Phylogenetic analysis of west nile virus genome, Iran. Emerg. Infect. Dis. 2014, 20, 1419. [Google Scholar] [CrossRef]
- Joosten, R.P.; Te Beek, T.A.; Krieger, E.; Hekkelman, M.L.; Hooft, R.W.; Schneider, R.; Sander, C.; Vriend, G. A series of pdb related databases for everyday needs. Nucleic Acids Res. 2010, 39, D411–D419. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Shahhosseini, N.; Chinikar, S.; Nowotny, N.; Fooks, A.R.; Schmidt-Chanasit, J. Genetic analysis of imported dengue virus strains by Iranian travelers. Asian Pac. J. Trop. Dis. 2016, 6, 850–853. [Google Scholar] [CrossRef]
- Doosti, S.; Yaghoobi-Ershadi, M.R.; Schaffner, F.; Moosa-Kazemi, S.H.; Akbarzadeh, K.; Gooya, M.M.; Vatandoost, H.; Shirzadi, M.R.; Mosta-Favi, E. Mosquito surveillance and the first record of the invasive mosquito species Aedes (stegomyia) albopictus (skuse)(diptera: Culicidae) in southern Iran. Iran. J. Public Health 2016, 45, 1064. [Google Scholar]
- Sedaghat, M.M.; Sarani, M.; Chinikar, S.; Telmadarraiy, Z.; Moghaddam, A.S.; Azam, K.; Nowotny, N.; Fooks, A.R.; Shahhosseini, N. Vector prevalence and detection of crimean-congo haemorrhagic fever virus in golestan province, Iran. J. Vector Borne Dis. 2017, 54, 353. [Google Scholar]
- Kayedi, M.H.; Sepahvand, F.; Mostafavi, E.; Chinikar, S.; Mokhayeri, H.; Sharafi, A.C.; Wong, G.; Shahhosseini, N.; Kazemi, S.H.M. Morphological and molecular identification of culicidae mosquitoes (diptera: Culicidae) in lorestan province, western Iran. Heliyon 2020, 6, e04480. [Google Scholar] [CrossRef]
- Bagheri, M.; Terenius, O.; Oshaghi, M.A.; Motazakker, M.; Asgari, S.; Dabiri, F.; Vatandoost, H.; Mohammadi Bavani, M.; Chavshin, A.R. West nile virus in mosquitoes of Iranian wetlands. Vector Borne Zoonotic Dis. 2015, 15, 750–754. [Google Scholar] [CrossRef]
- Jupp, P.; McIntosh, B.; Dickinson, D. Quantitative experiments on the vector capability of culex (culex) theileri theobald with west nile and sindbis viruses. J. Med. Entomol. 1972, 9, 393–395. [Google Scholar] [CrossRef]
- Meshkat, Z.; Chinikar, S.; Shakeri, M.; Manavifar, L.; Moradi, M.; Mirshahabi, H.; Jalali, T.; Khakifirouz, S.; Shahhosseini, N. Prevalence of west nile virus in mashhad, Iran: A population–based study. Asian Pac. J. Trop. Med. 2015, 8, 203–205. [Google Scholar] [CrossRef]
- Fereidouni, S.R.; Ziegler, U.; Linke, S.; Niedrig, M.; Modirrousta, H.; Hoffmann, B.; Groschup, M.H. West nile virus monitoring in migrating and resident water birds in Iran: Are common coots the main reservoirs of the virus in wetlands? Vector Borne Zoonotic Dis 2011, 11, 1377–1381. [Google Scholar] [CrossRef]
- Ziyaeyan, M.; Behzadi, M.A.; Leyva-Grado, V.H.; Azizi, K.; Pouladfar, G.; Dorzaban, H.; Ziyaeyan, A.; Salek, S.; Hashemi, A.J.; Jamalidoust, M. Widespread circulation of west nile virus, but not zika virus in southern Iran. PLoS Negl. Trop. Dis. 2018, 12, e0007022. [Google Scholar] [CrossRef]
- Molaei, G.; Andreadis, T.G.; Armstrong, P.M.; Anderson, J.F.; Vossbrinck, C.R. Host feeding patterns of culex mosquitoes and west nile virus transmission, northeastern united states. Emerg. Infect. Dis 2006, 12, 468. [Google Scholar] [CrossRef]
- Burkett-Cadena, N.D.; Graham, S.P.; Hassan, H.K.; Guyer, C.; Eubanks, M.D.; Katholi, C.R.; Unnasch, T.R. Blood feeding patterns of potential arbovirus vectors of the genus culex targeting ectothermic hosts. Am. J. Trop. Med. Hyg. 2008, 79, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Fritz, M.; Walker, E.; Miller, J.; Severson, D.; Dworkin, I. Divergent host preferences of above—And below—Ground c ulex pipiens mosquitoes and their hybrid offspring. Med. Vet. Entomol. 2015, 29, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Brugman, V.; Hernández-Triana, L.; England, M.; Medlock, J.; Mertens, P.P.; Logan, J.; Wilson, A.; Fooks, A.; Johnson, N.; Carpenter, S. Blood-feeding patterns of native mosquitoes and insights into their potential role as pathogen vectors in the thames estuary region of the united kingdom. Parasit. Vectors 2017, 10, 163. [Google Scholar] [CrossRef]
- May, F.J.; Davis, C.T.; Tesh, R.B.; Barrett, A.D. Phylogeography of west nile virus: From the cradle of evolution in africa to eurasia, australia, and the americas. J. Virol. 2011, 85, 2964–2974. [Google Scholar] [CrossRef] [PubMed]
- Olsen, B.; Munster, V.J.; Wallensten, A.; Waldenström, J.; Osterhaus, A.D.; Fouchier, R.A. Global patterns of influenza a virus in wild birds. Science 2006, 312, 384–388. [Google Scholar] [CrossRef]
- Ergunay, K.; Bakonyi, T.; Nowotny, N.; Ozkul, A. Close relationship between west nile virus from turkey and lineage 1 strain from central african republic. Emerg. Infect. Dis. 2015, 21, 352. [Google Scholar] [CrossRef]
- Botha, E.M.; Markotter, W.; Wolfaardt, M.; Paweska, J.T.; Swanepoel, R.; Palacios, G.; Nel, L.H.; Venter, M. Genetic determinants of virulence in pathogenic lineage 2 west nile virus strains. Emerg. Infect. Dis. 2008, 14, 222. [Google Scholar] [CrossRef]
- Papa, A.; Bakonyi, T.; Xanthopoulou, K.; Vázquez, A.; Tenorio, A.; Nowotny, N. Genetic characterization of west nile virus lineage 2, greece, 2010. Emerg. Infect. Dis. 2011, 17, 920. [Google Scholar] [CrossRef]
- McMullen, A.R.; Albayrak, H.; May, F.J.; Davis, C.T.; Beasley, D.W.; Barrett, A.D. Molecular evolution of lineage 2 west nile virus. J. Gen. Virol. 2013, 94, 318. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Ray, D.; Zhao, Y.; Dong, H.; Ren, S.; Li, Z.; Guo, Y.; Bernard, K.A.; Shi, P.-Y.; Li, H. Structure and function of flavivirus ns5 methyltransferase. J. Virol. 2007, 81, 3891–3903. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–62. [Google Scholar] [PubMed]
- Murata, R.; Eshita, Y.; Maeda, A.; Maeda, J.; Akita, S.; Tanaka, T.; Yoshii, K.; Kariwa, H.; Umemura, T.; Takashima, I. Glycosylation of the west nile virus envelope protein increases in vivo and in vitro viral multiplication in birds. Am. J. Trop. Med. Hyg. 2010, 82, 696–704. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mosquito Species | No. of Mosquitoes/Species and (%) of Total | No. of Blood-Fed Mosquitoes/Species and (%) of Total | No. of Blood-Fed Mosquitoes/Host and (%) of Total | |||||
|---|---|---|---|---|---|---|---|---|
| Homo Sapiens | Sus Scrofa | Bos Taurus | Canis Lupus | Ovis Aries | Anas Sparsa | |||
| Cx. pipiens Co. | 2885 (57.4) | 8 (10.5) | 5 (23.8) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (42.9) |
| Cx. theileri | 1224 (24.3) | 31 (40.8) | 13 (61.9) | 13 (72.2) | 3 (18.8) | 2 (18.2) | 0 (0) | 0 (0) |
| Cx. perexiguus | 163 (3.2) | 11 (14.5) | 3 (14.3) | 3 (16.7) | 0 (0) | 1 (9.1) | 0 (0) | 4 (57.1) |
| An. sacharovi | 535 (10.6) | 4 (5.3) | 0 (0) | 1 (5.6) | 1 (6.3) | 2 (18.2) | 0 (0) | 0 (0) |
| An. stephensi | 83 (1.7) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| An. superpictus | 75 (1.5) | 2 (2.6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 2 (66.7) | 0 (0) |
| An. dthali | 32 (0.6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| An. maculipennis | 28 (0.6) | 20 (26.3) | 0 (0) | 1 (5.6) | 12 (75) | 6 (54.5) | 1 (33.3) | 0 (0) |
| An. fluviatilis | 3 (0.1) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| Total | 5028 (100) | 76 (100) | 21 | 18 | 16 | 11 | 3 | 7 |
| Gene | Strain/Subclade/Pathogenicity | Iran (MN238670)/2g | Iran (MN238669)/2g | Russia (FJ425721)/2g/high | SA (EF429200) /2f/high | SA (EF429199)/2f/less | CAR (DQ318020)/2f/high | MAD (HM147823)/2e/attenuated | |
|---|---|---|---|---|---|---|---|---|---|
| a.a Position | |||||||||
| C | - | - | - | - | - | - | - | - | |
| prM | 73 | P | - | - | S | S | S | S | |
| 91 | - | - | - | - | - | K | - | ||
| 105 | A | - | - | - | V | - | - | ||
| 221 | A | - | - | G | - | - | - | ||
| E | 66 | S | - | - | N | - | - | - | |
| 70 | T | - | - | P | - | - | - | ||
| 71 | K | - | - | R | R | R | - | ||
| 154 | N | - | - | - | - | D | - | ||
| 159 | M | - | - | I | I | I | V | ||
| 250 | Q | - | - | - | - | R | - | ||
| 312 | A | - | - | - | V | - | - | ||
| NS1 | 88 | V | - | - | - | I | I | - | |
| 98 | K | - | - | - | - | R | - | ||
| 290 | S | - | - | - | R | - | - | ||
| 338 | T | - | - | - | - | - | L | ||
| 401 | I | - | V | - | - | - | - | ||
| 442 | L | - | M | - | - | - | - | ||
| NS2A | 126 | S | - | - | - | - | - | L | |
| 190 | K | - | - | R | - | - | - | ||
| 259 | S | - | - | T | - | - | - | ||
| NS2B | - | - | - | - | - | - | - | - | |
| NS3 | 160 | S | - | - | - | A | - | A | |
| 298 | R | - | - | - | G | - | - | ||
| 333 | I | - | - | - | V | - | - | ||
| 421 | S | - | - | - | - | - | N | ||
| 572 | T | - | - | M | - | - | - | ||
| 597 | A | - | - | V | - | - | - | ||
| NS4A | 98 | E | - | - | - | - | D | - | |
| 100 | P | - | S | - | - | - | - | ||
| NS4B | 20 | P | - | - | - | - | - | L | |
| 117 | A | - | - | V | - | - | - | ||
| 274 | H | Y | Y | - | - | - | - | ||
| NS5 | 42 | H | - | - | - | Q | - | - | |
| 117 | K | - | - | - | R | - | - | ||
| 254 | F | - | - | - | - | - | Y | ||
| 278 | K | - | - | - | R | - | - | ||
| 281 | N | - | - | - | S | - | - | ||
| 370 | E | - | A | - | - | - | - | ||
| 403 | R | - | - | G | G | G | - | ||
| 528 | P | - | - | - | G | - | S | ||
| 816 | A | - | - | S | - | S | - | ||
| 837 | K | - | - | R | - | R | - | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahhosseini, N.; Moosa-Kazemi, S.H.; Sedaghat, M.M.; Wong, G.; Chinikar, S.; Hajivand, Z.; Mokhayeri, H.; Nowotny, N.; Kayedi, M.H. Autochthonous Transmission of West Nile Virus by a New Vector in Iran, Vector-Host Interaction Modeling and Virulence Gene Determinants. Viruses 2020, 12, 1449. https://doi.org/10.3390/v12121449
Shahhosseini N, Moosa-Kazemi SH, Sedaghat MM, Wong G, Chinikar S, Hajivand Z, Mokhayeri H, Nowotny N, Kayedi MH. Autochthonous Transmission of West Nile Virus by a New Vector in Iran, Vector-Host Interaction Modeling and Virulence Gene Determinants. Viruses. 2020; 12(12):1449. https://doi.org/10.3390/v12121449
Chicago/Turabian StyleShahhosseini, Nariman, Seyed Hassan Moosa-Kazemi, Mohammad Mehdi Sedaghat, Gary Wong, Sadegh Chinikar, Zahra Hajivand, Hamid Mokhayeri, Norbert Nowotny, and Mohammad Hassan Kayedi. 2020. "Autochthonous Transmission of West Nile Virus by a New Vector in Iran, Vector-Host Interaction Modeling and Virulence Gene Determinants" Viruses 12, no. 12: 1449. https://doi.org/10.3390/v12121449
APA StyleShahhosseini, N., Moosa-Kazemi, S. H., Sedaghat, M. M., Wong, G., Chinikar, S., Hajivand, Z., Mokhayeri, H., Nowotny, N., & Kayedi, M. H. (2020). Autochthonous Transmission of West Nile Virus by a New Vector in Iran, Vector-Host Interaction Modeling and Virulence Gene Determinants. Viruses, 12(12), 1449. https://doi.org/10.3390/v12121449