1. Introduction
The baculovirus expression vector system (BEVS) is one of the most versatile platform technologies developed in the past 30 years to produce complex (glyco)proteins in eukaryotic cells, because it frequently results in high yields of purified proteins and allows for similar post-translational modifications as mammalian expression systems [
1]. An increasing number of commercial BEVS products are becoming available, and these include human vaccines against influenza A virus and human papilloma virus, gene therapy products, and veterinary vaccines [
2,
3]. More BEVS bio-pharmaceuticals are currently in (pre)clinical trials and these include prototype COVID-19 vaccines that are aimed at protection against SARS-CoV-2 infection [
4]. Traditional baculovirus expression vectors are generated by homologous recombination in insect cells between wildtype baculovirus DNA and a so-called transfer vector (plasmid DNA), which carries the gene of interest (GOI) under a suitable baculovirus promoter, typically the very strong polyhedrin or p10 promoter [
5,
6]. There have been sophisticated improvements throughout the years to streamline the generation and isolation of recombinant baculoviruses, and numerous easy-to-use commercial baculovirus kits are available [
7].
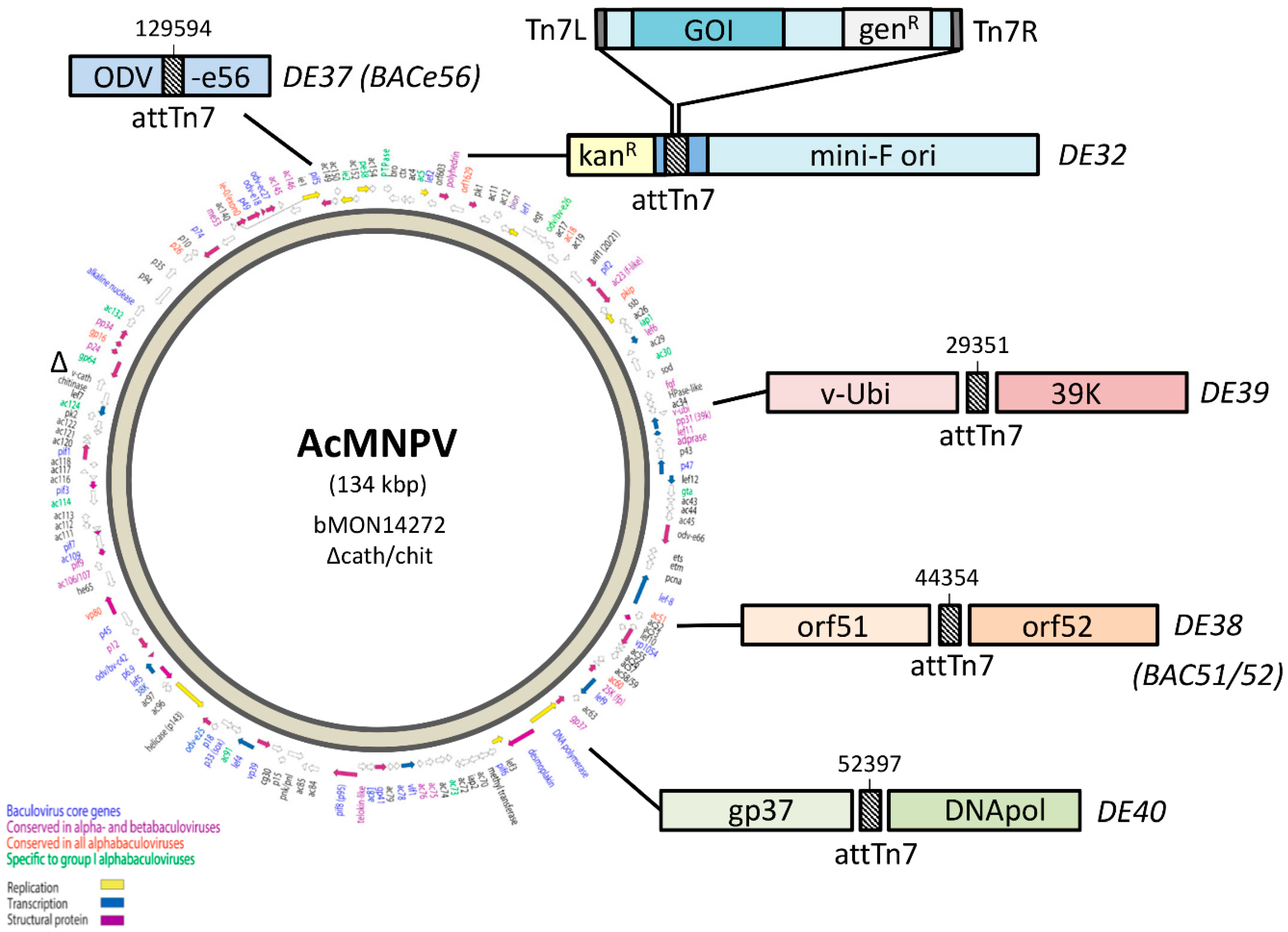
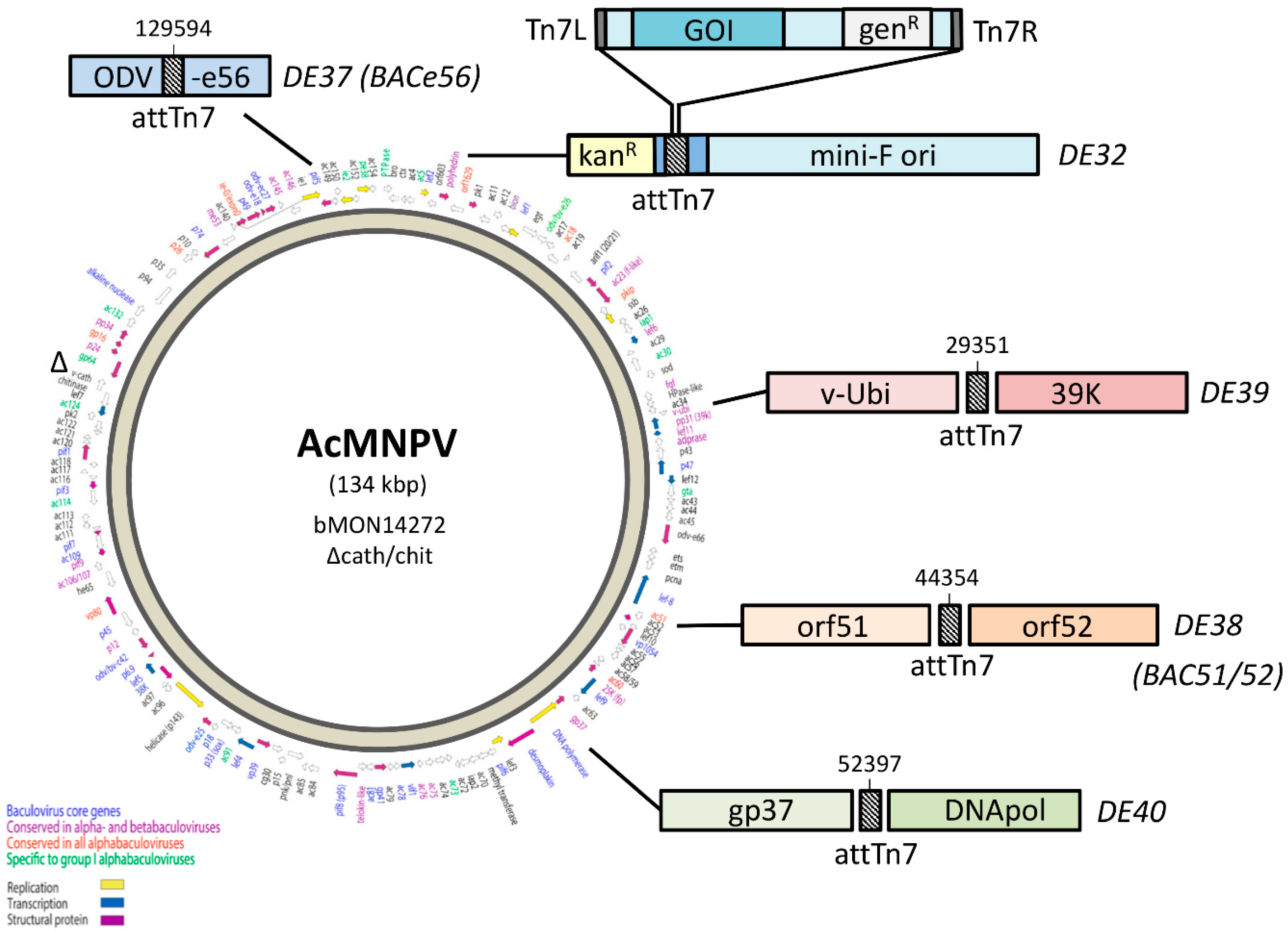
One of the major breakthroughs in baculovirus technology was the generation of an
Autographa californica multiple nucleopolyhedrovirus (AcMNPV) infectious clone that could be maintained as a ~140 kb large, single-copy bacterial artificial chromosome (BAC) in
E. coli [
8]. This so-called “bacmid” carries a mini-F replicon for single-copy replication in
E. coli, a kanamycin resistance gene (kan
R) for selection, and a LacZ-attTn7 transposon integration site (inserted between kan
R and mini-F) for insertion of the GOI by Tn7 transposition (
Figure 1). A commercial kit based on these bacmids was successfully launched by Invitrogen Life Technologies (now ThermoFisher) as the “Bac-to-Bac baculovirus expression system”, which uses so-called “pFastBac” transfer vectors in which the GOI is cloned in between attL and attR sites, and downstream of the strong polyhedrin (polh) or p10 promoter. After Tn7 transposition, the recombinant bacmids are selected for antibiotic resistance (kan
R and gen
R) and, upon transfection of insect cells, this yields, at least in theory, a genetically homogeneous recombinant baculovirus stock that does not require further plaque purification.
While bacmids are efficient for rapidly creating recombinant baculoviruses, the introduction of genes is limited to a single insertion site. MultiBac (Geneva Biotech) has improved the system to allow for the introduction of several genes, with the multigene construct being shuttled into the same attTn7 site [
9]. The original AcMNPV bacmid (bMON14272, [
8]) can be easily manipulated in
E. coli and this has been the driving force for initiating numerous knock out studies to study baculovirus gene functions. Bacmids have been constructed for baculoviruses other than AcMNPV [
10,
11,
12] and this has tremendously accelerated functional studies on previously uncharacterized baculovirus genes, including baculovirus core genes or those involved in oral infectivity [
13,
14,
15,
16].
Despite the major contribution of bacmids in the understanding of baculovirus biology, most commercial baculovirus vectors are still based on the traditional method of homologous recombination in insect cells between a transfer plasmid carrying the GOI and a (linearized) AcMNPV genome, followed by several rounds of plaque purification and master seed production.
For large-scale protein expression, bacmid-based expression vectors have not been applied, despite improvements that have increased the efficiency of GOI transposition and recombinant bacmid selection [
17]. This may change soon, however, since Novavax appears to produce its COVID-19 vaccine NVX-CoV2373 using a pFastBac vector containing a SARS-CoV-2 spike gene [
18]. An important reason for not using bacmids for large-scale production is the relative rapid loss of GOI expression upon serial passage [
19] resulting from the genomic instability of the mini-F replicon in the bacmid backbone [
20]. The exact reason for the rapid deletion of the mini-F and the adjacent GOI is unknown, but its bacterial origin and large size (>8 kbp) likely account for a negative selection pressure on recombinant baculovirus genomes during replication in insect cells.
We hypothesize that relocation of the attTn7 away from the mini-F replicon would prevent or at least delay GOI deletion, thereby resulting in higher recombinant protein expression levels upon serial passage. To test this hypothesis, we generate a series of bacmids with attTn7 insertions at different locations on the AcMNPV genome that had previously been shown to tolerate insertions [
21]. We analyze the performance of the different bacmids by comparing the relative expression levels of different GOIs. The best performing bacmid is subsequently tested for the stability of recombinant protein expression upon serial undiluted passages in insect cells. Finally, we demonstrate the viability of this novel bacmid for industrial vaccine scale-up by producing chikungunya virus-like particles (CHIKV VLPs) in Sf9 suspension cells in shaker flasks and a bioreactor [
22,
23,
24,
25].
2. Materials and Methods
2.1. Construction of ΔattTn7 Bacmid
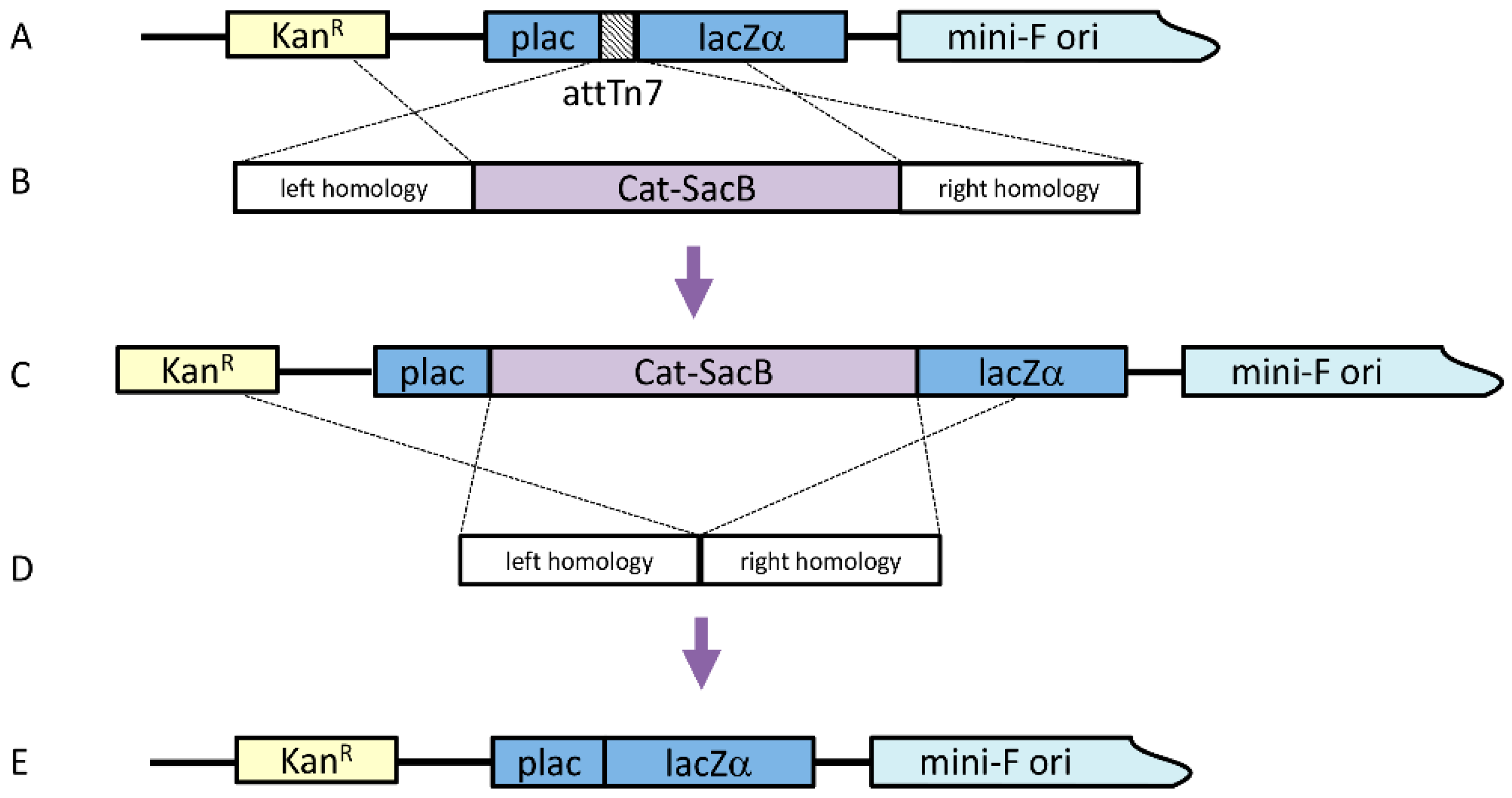
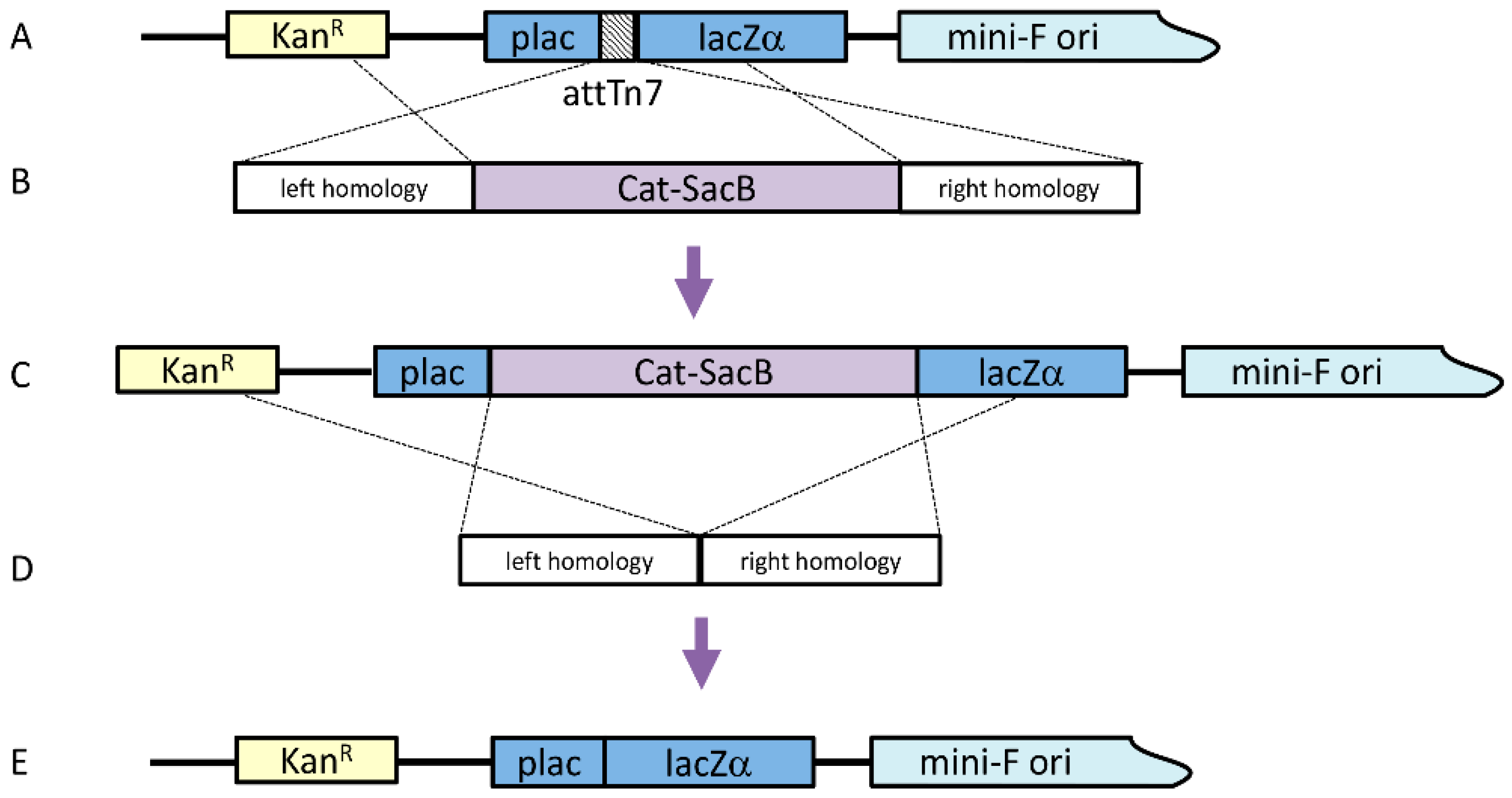
Before moving the attTn7 binding region to alternate locations, the original attTn7 site had to be removed. To knock out the attTn7 binding region, a linear cassette was generated by a Gibson Isothermal Assembly, which included ~300 bp homology arms in both the pLac and LacZ regions along with the counter-selectable cassette Cat-SacB (chloramphenicol acetyltransferase-sucrose sensitivity), which was amplified from pELO4 (gift from Don Court, NCI); both homology arms included 40 bp of sequence homology at the appropriate ends of the Cat-SacB cassette. Twenty-five femtomoles of each amplicon were included in a 20 μL reaction with 2× Gibson Assembly Master Mix (New England Biolabs, Ipswich, MA, USA) and incubated at 50 °C for 30 min. To amplify the assembled cassette, 1 μL of the assembly reaction was used as a template in a 100 μL PCR reaction, including 0.4 μM of each primer and 2× Phusion HF Mastermix (60 °C annealing temperature, 4 min extension, 25 cycles). The linear cassette was purified using a QiaQuick PCR Purification Kit (Qiagen, Hilden, Germany).
The original strain for this work was λRed E. coli SW106 (gift from Don Court, NCI) harboring an AcMNPV bacmid, from which the chitinase and cathepsin genes had previously been deleted (bMON14272Δvcath-chiA or DE32). The strain was grown in LB with 50 μg/mL kanamycin at 30 °C with shaking to OD600 of 0.4. The culture was agitated in a 42 °C water bath for 15 min to induce the temperature-sensitive lambda prophage. The cells were then chilled and made electrocompetent. One hundred nanograms of the knock-out cassette were added to 50 μL of the cells, which were subsequently electroporated. The transformation was incubated with shaking for two hours at 30 °C in 1 mL LB before plating 100 μL on LB agar with 50 μg/mL kanamycin and 20 μg/mL chloramphenicol at 30 °C. Four colonies were selected on the second morning and diluted in 50 μL water, with 1 μL being used as a template for colony PCR with primers that bound just outside the insertion.
Once this intermediate bacmid was confirmed, the Cat-SacB selection cassette was seamlessly removed. Similar ~300 bp arms, homologous to the end of pLac and the beginning of LacZ, were generated by PCR, where the LacZ arm product included 40 bp homology to the end of pLac. The two amplicons were assembled by overlap PCR (0.2 μL of each amplicon, 2× HF mastermix, 0.4 μM primers, 100 μL total volume) (60 °C annealing, 30 s extension, 25 cycles) to produce the marker knock-out cassette and were purified using a QiaQuick PCR Purification Kit. Lambda Red was induced in the SW106 bMON14272Δvcath-chiAΔattTn7:Cat-SacB strain and electrocompetent cells were made as described above. One hundred nanograms of the marker removal cassette was added to 50 μL of electrocompetent cells and transformed by electroporation. The cells were allowed to recover by shaking at 30 °C for 4 h in 10 mL LB. Cells from 1 mL of the transformation were pelleted by centrifugation for 30 s at 13,000× g and resuspended in 1 volume 1× M9 salts twice to wash the cells. After the second wash, the cells were resuspended in 100 µl M9 salts and plated on LB agar with no salt, 6% sucrose, and 50 µg/mL kanamycin at 37 °C, to select cells that no longer contained the Cat-SacB selection marker. Bacmid DNA was isolated from four colonies by alkaline lysis and the recombined region was amplified by PCR to confirm the deletion of the Cat-SacB marker. The resulting bacmid with the correctly sized amplicon was verified by sequencing the entire cloned region.
The confirmed bMON14272Δvcath-chiAΔ
attTn7 was used to transform
E. coli DE25 cells, a DH10B-derived cell line compatible with the Bac-to-Bac system for recombinant baculovirus production. The strain contains an optimized helper plasmid that encodes Tn7 transposition functions to generate recombinant baculovirus and confers resistance to tetracycline [
17]. The transformation was plated on LB with 50 µg/mL kanamycin and 12.5 µg/mL tetracycline at 37 °C. A single colony was selected from the plate to generate competent cells for the new deletion strain.
2.2. Construction of Bacmids with Relocated attTn7
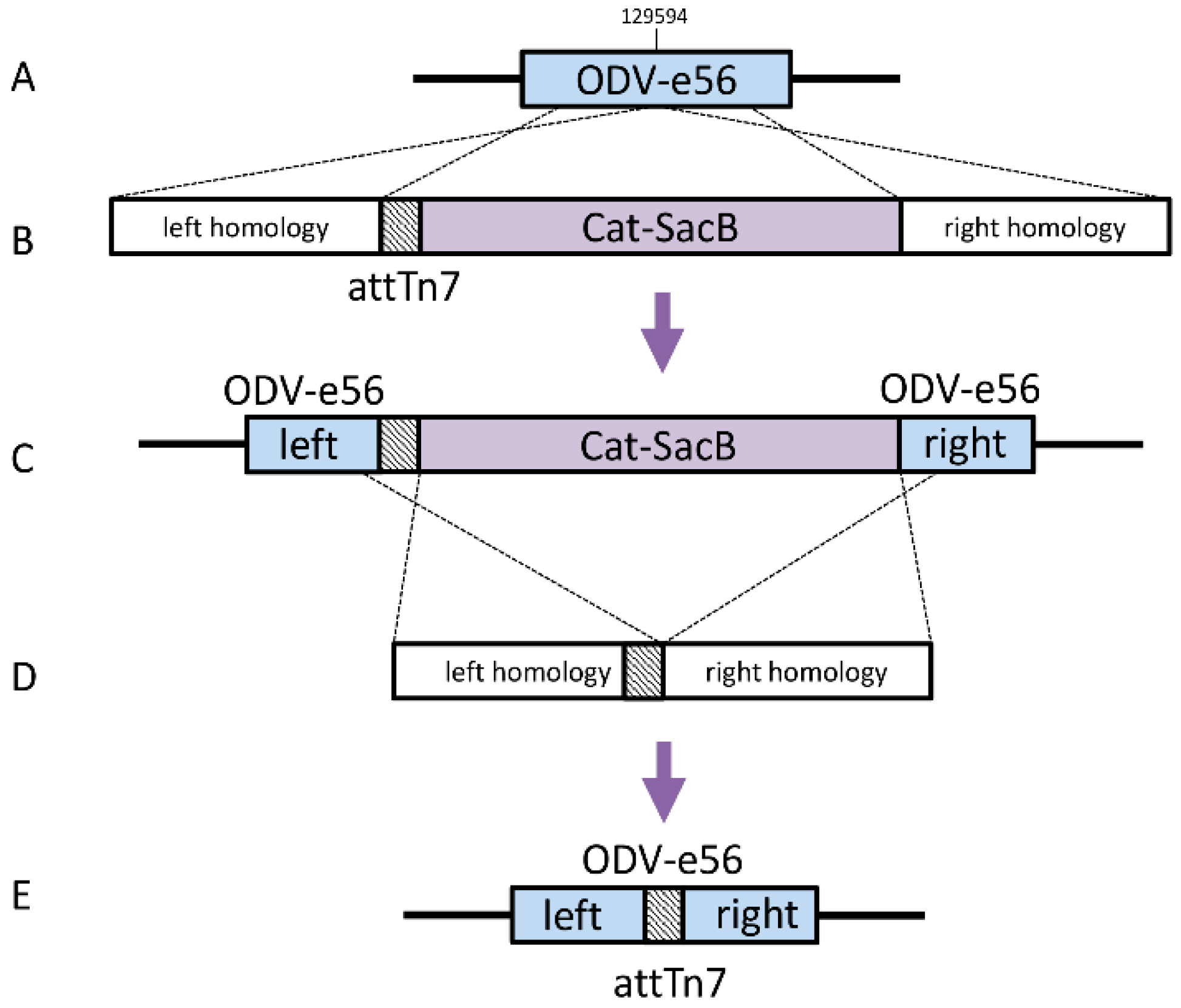
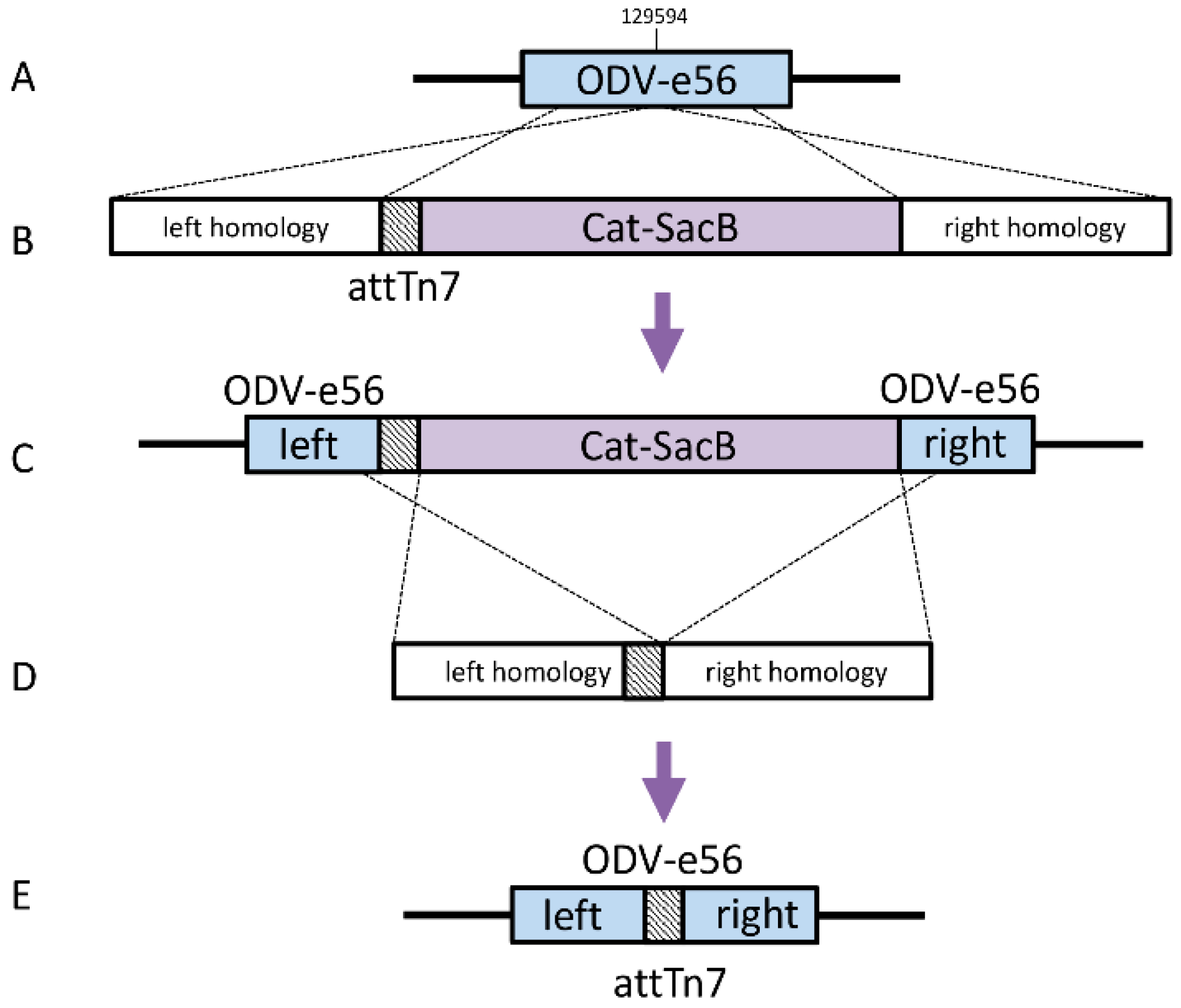
Bacmids with relocated attTn7 sites Strains DE37, DE38, DE39, and DE40 were generated in a similar fashion. The knock-in cassettes for odv-e56 (pif-5) and the intergenic regions of orf51/52, v-ubiquitin (v-ubi)/39 k, and gp37/DNApol included 4 amplicons, which contained a ~300 bp region of homology to the immediate left of the desired insertion point with a 40 bp homology to attTn7 (Forward: 5′- TGTGGAATTGTGAGCGGATA; Reverse: 5′-TCCTGTGACGGAAGATCACTTCGCAGAATAAAT AAATCCTGGTGCTGCAAGGCGATTAAGT), Cat-SacB cassette, and ~300b p region of homology to the right of the insertion including 40 bp homology to the end of SacB (
Table 1).
The amplicons were assembled by Isothermal Assembly with 25 femtomoles of each piece in a 20 μL reaction with 2× Gibson Assembly Master Mix (New England Biolabs) and incubated for 30 min at 50 °C. The cassette was then amplified with 1 μL of the assembly reaction as a template in a 100 uL PCR reaction, including 0.4 μM of each primer and 2× Phusion HF Mastermix (60 °C annealing temperature, 4 min extension, 25 cycles). The linear cassette was purified using a QiaQuick PCR Purification Kit (Qiagen).
The previously generated strain, SW106 bMON14272Δvcath-chiA; ΔattTn7, was induced as described previously and electrocompetent cells were made. One hundred nanograms of the attTn7 knock-in cassette was added to 50 μL of electrocompetent cells and electroporated at 1.8 kV. The cells were recovered in 1 mL LB at 30 °C for 2 h and 100 μL was then plated on LB with 50 ug/mL kanamycin and 20 ug/mL chloramphenicol. Four colonies were selected on the second morning and diluted in 50 uL water, with 1ul being used as template for colony PCR, with primers landing just outside the insertion. The remaining diluted cells were then used to seed cultures to generate glycerol stocks for the selected positive construct. The Cat-SacB marker was removed from these intermediate strains in a manner similar to that described above. The loss of the marker was confirmed by PCR amplification of the insertion region and sequencing of the entire region for each new bacmid.
The confirmed bacmids were transformed into E. coli DE25 cells as described above. A single colony was selected from each plate to generate competent cells for DE37, DE38, DE39, and DE40. The entire bacmid for each strain was analyzed by PacBio sequencing to be certain that there was no unintended rearrangement during recombination.
2.3. Generation of Recombinant Baculoviruses
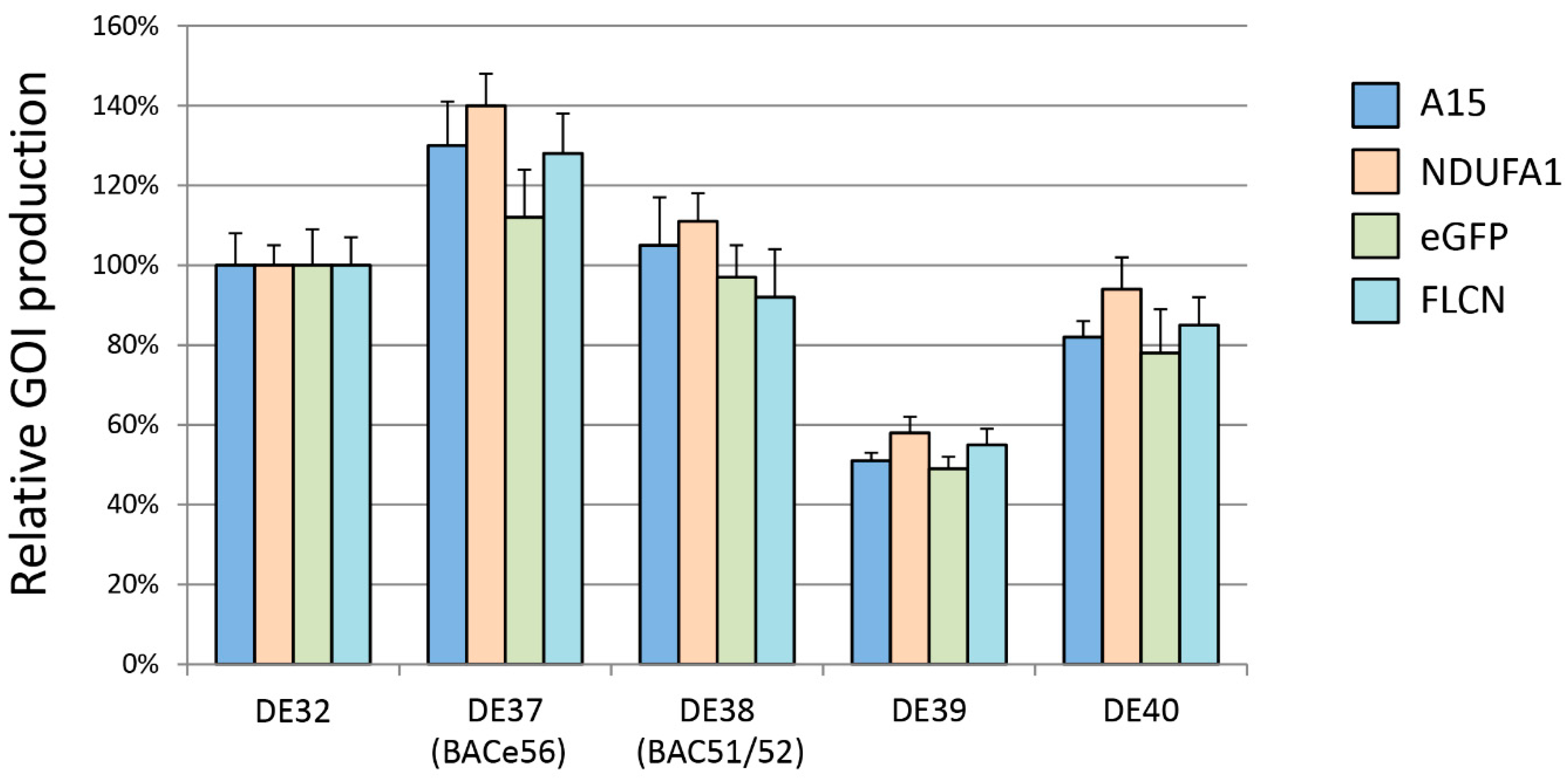
To test the expression levels at each location, four GOIs were tested: mouse A15 extracellular domain (NP_062608.2), human NDUFA13 (NP_057049.5), human FLCN (NP_659434.2), and enhanced green fluorescent protein (eGFP). Expression clones were generated for each by gateway recombination of previously validated entry clones into baculovirus expression backbones (pDest-636), including the polh promoter and N-terminal His6-MBP (maltose-binding protein) tag. To facilitate shuttling into the bacmid, the expression backbone contained Tn7 left and right arms as well as the gentamycin resistance marker. One microliter of the verified expression clone was used to transform 50 μL of chemically competent cells of each of the new strains (DE37-DE40) as well as the DE32 (Δvcath-chiA) control. One ml of LB was added to the transformants and cultures were allowed to grow at 37 °C for 4 h with shaking. Ten microliters of the outgrowth were diluted into 190 μL LB and the resulting 200 μL was plated onto LB agar plates with gentamycin (7 ug/mL), kanamycin (50 ug/mL), tetracycline (10 ug/mL), IPTG (40 ug/mL), and Bluo-gal (100 ug/mL), and grown overnight at 37 °C. White colonies were selected from each plate and grown overnight in 3 mL LB with gentamycin and kanamycin at 37 °C with shaking. Two milliliter aliquots were pelleted and bacmid DNA was prepared by alkaline lysis. Junction PCR was performed to verify the correct inserts using the lysis as template. Fifty milliliters of Sf9s at a density of 1.5 × 106 cells/mL was transfected with 25 μL of bacmid complexed with 125 μL of Insect GeneJuice (Sigma-Aldrich, Saint Louis, MO, USA). The complexes were allowed to form for 20 min and were then added to the culture. The cultures were incubated for 5 days and then harvested by centrifugation at 2000 RPM for 10 min and the supernatants were then collected in 50 mL conical tubes.
2.4. Recombinant Protein Production and Quantitation
For small-scale protein expression, 50 mL of Tni-FNL cells were seeded at 1.5 × 106 cells/mL and were infected with the generated viruses at a multiplicity of infection (MOI) of 3. The infected cultures were allowed to grow at 27 °C for 72 h. The cells were then harvested by centrifugation at 2500 rpm for 20 min in 50 mL conical tubes. The pellets were each resuspended in 5 mL buffer (20 mM Hepes pH 7.3, 300 mM NaCl, 1 mM TCEP) by gently vortexing, and then were lysed using an LV-1 microfluidizer (Microfluidics, Inc., Westwood, MA, USA) set at a pressure of 7000 psi for 2 passes per lysate. Lysates were then clarified using ultracentrifugation at 33,100 rpm for 30 min, and the supernatant was collected in a 15 mL conical tube. Clarified cell extracts of A15, NDUFA13, and FLCN were purified using immobilized metal affinity chromatography on Phynexus tips with elution in 500 mM imidazole. Final proteins were analyzed by SDS–PAGE chromatography to ensure purity, and quantitated in a Nanodrop spectrophotometer at 280 nm. Cell extracts expressing eGFP were quantified by measuring GFP fluorescence (excitation = 485 nm, emission = 510 nm) on a BMG Omega plate reader. Data presented are averages of three replications of the purification process with the standard deviation shown via error bars.
2.5. Serial Undiluted Baculovirus Passage on Sf21 Cells
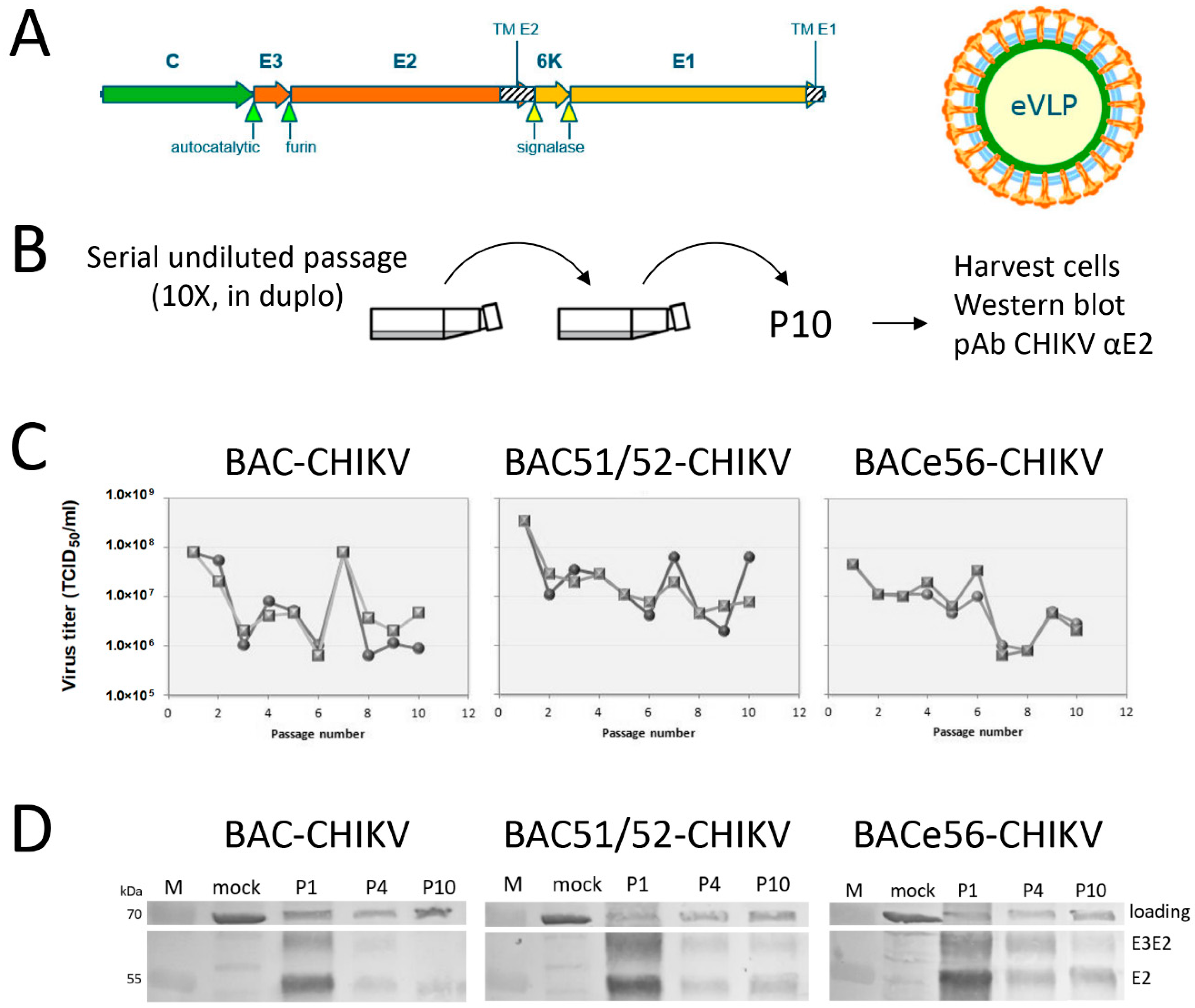
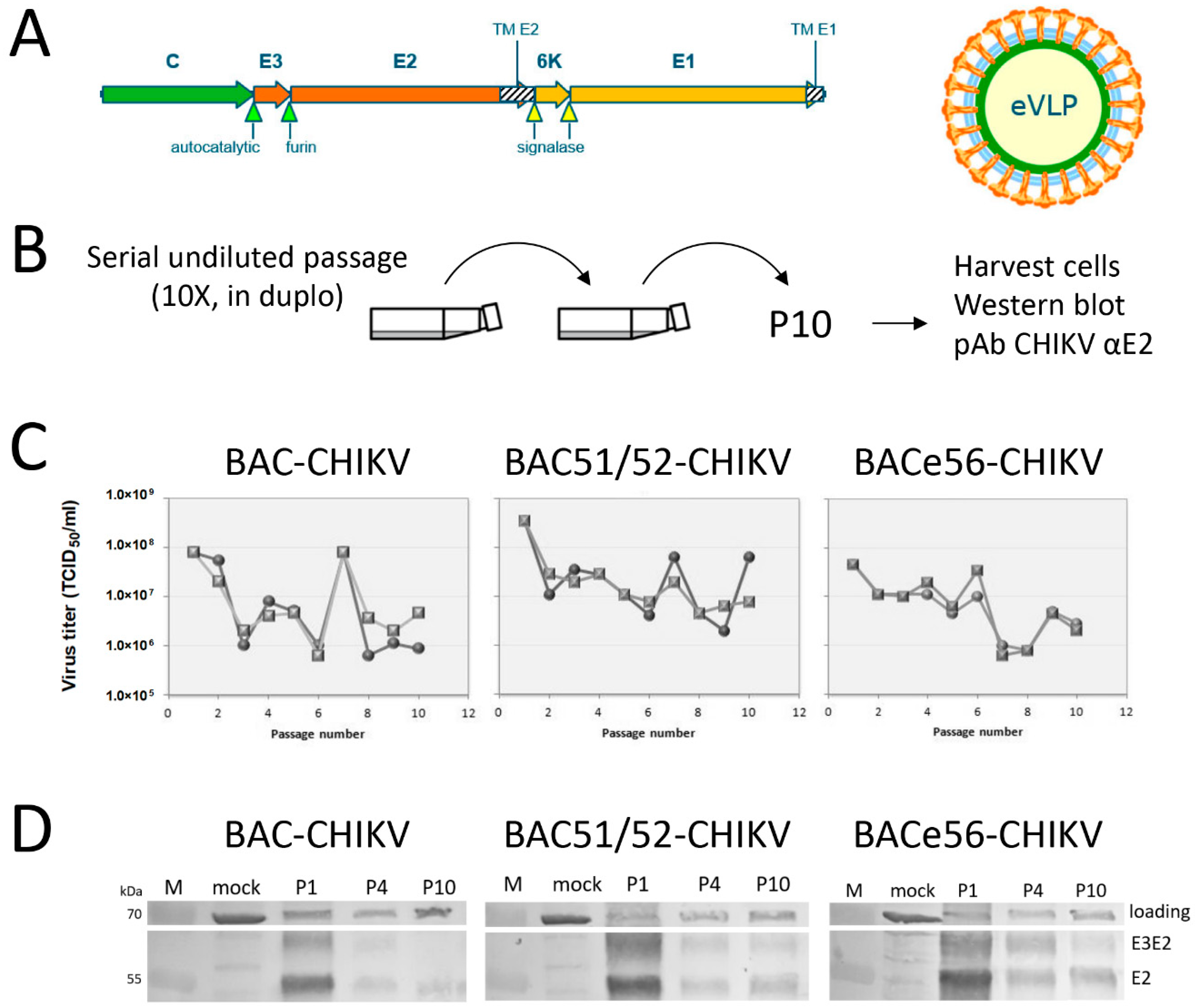
Bacmid-derived viruses expressing CHIKV VLPs were passaged ten times in duplicate in Sf21 cells, as described before [
19,
25]. Sf21 cells were cultured in supplemented Grace’s insect medium (Gibco, Life Technologies, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA) and 50 μg/mL gentamycin (Gibco, Life Technologies) in a T25 cell culture flask (Greiner, Alphen aan den Rijn, the Netherlands). For serial passage, 1 mL of each virus suspension was added to a T25 flask that contained healthy Sf21 cells with a confluency of 50–60%. After three hours, the virus suspension was replaced with 4 mL fresh medium. The cells were incubated for 3 days at 27 °C. Cells were detached and the cell suspension was centrifuged for 5 min at 4000 rpm. The cell pellet was washed once with 500 µL PBS and resuspended in PBS with protease inhibitor (Roche, Basel, Switserland). For the next virus passage, 1 mL of the supernatant was added to new healthy Sf-21 cells. The remaining supernatant was stored at 4 °C. This procedure was repeated ten times in duplicate for each virus.
2.6. SDS–PAGE and Western Blot Immunodetection
Protein samples containing equal numbers of cells were analyzed in 12.5% SDS–PAGE gels (Mini-PROTEAN® Tetra System, Bio-Rad Laboratories, Hercules, CA, USA). Proteins were transferred to an Immobilon-P membrane (Millipore) using a Tris–Glycine buffer (25 mM Tris, 192 mM glycine, 10% (v/v) methanol, pH 8.3). Membranes were blocked in 3% low-fat milk powder (Campina, The Netherlands) in PBS 0.1% (v/v) Tween-20 (PBS-T) (Merck). Blots were washed with 2.5 mL PBS-T for 5 min at RT and the primary polyclonal antibody rabbit-anti-CHIKV-E2 or -E1 immunoglobulin was added (dilution 1:10,000). For detection of the AcMNPV VP39 major capsid protein, a polyclonal mouse anti-VP39 immunoglobulin was used (dilution 1:1000). Secondary antibodies were polyclonal goat anti-rabbit immunoglobulin (AP-conjugated) or polyclonal goat anti-mouse immunoglobulin (AP-conjugated) (both Sigma-Aldrich). After incubation and washing, the blot was stained with NBT/BCIP (Roche Diagnostics GmbH, Basel, Switserland).
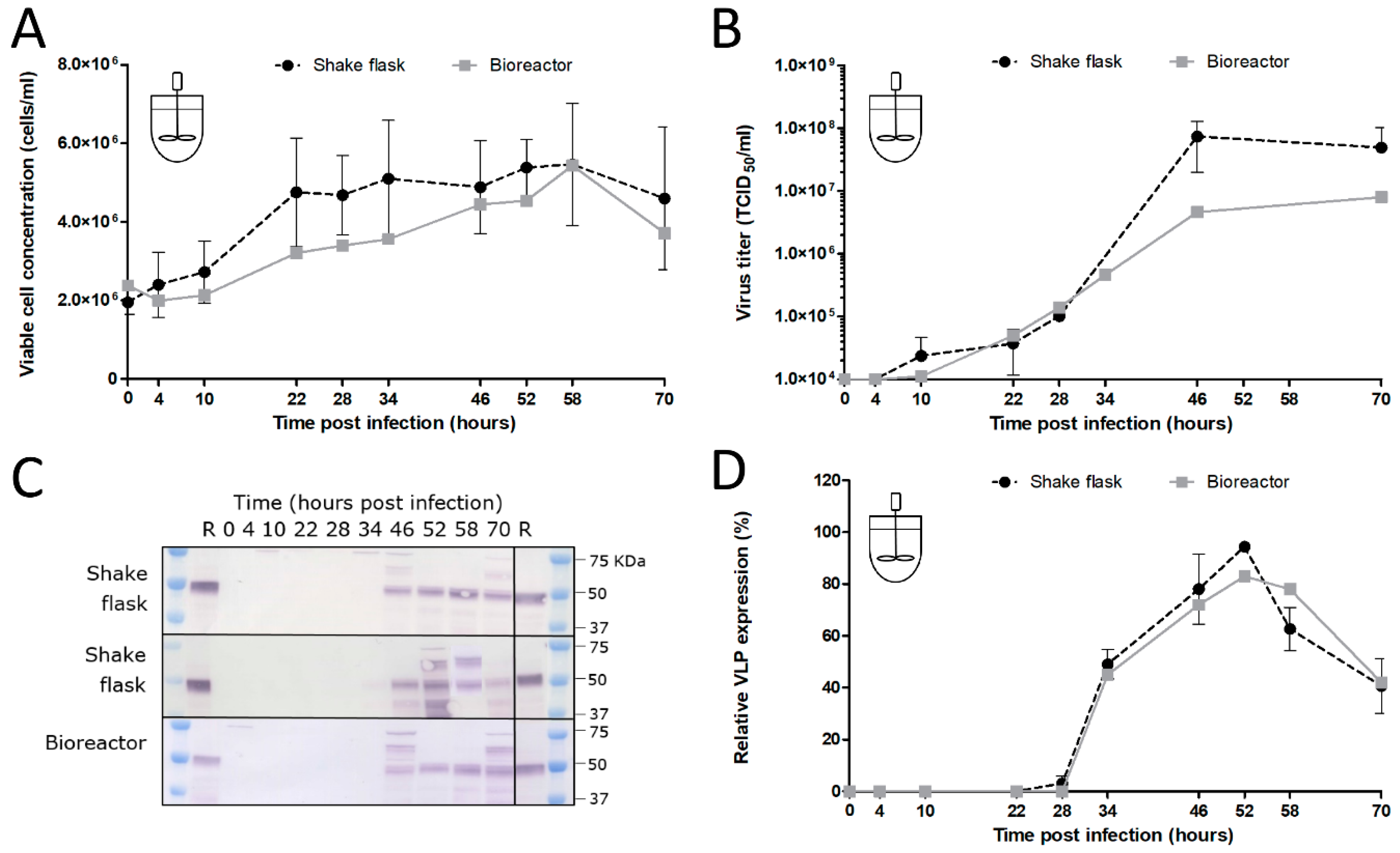
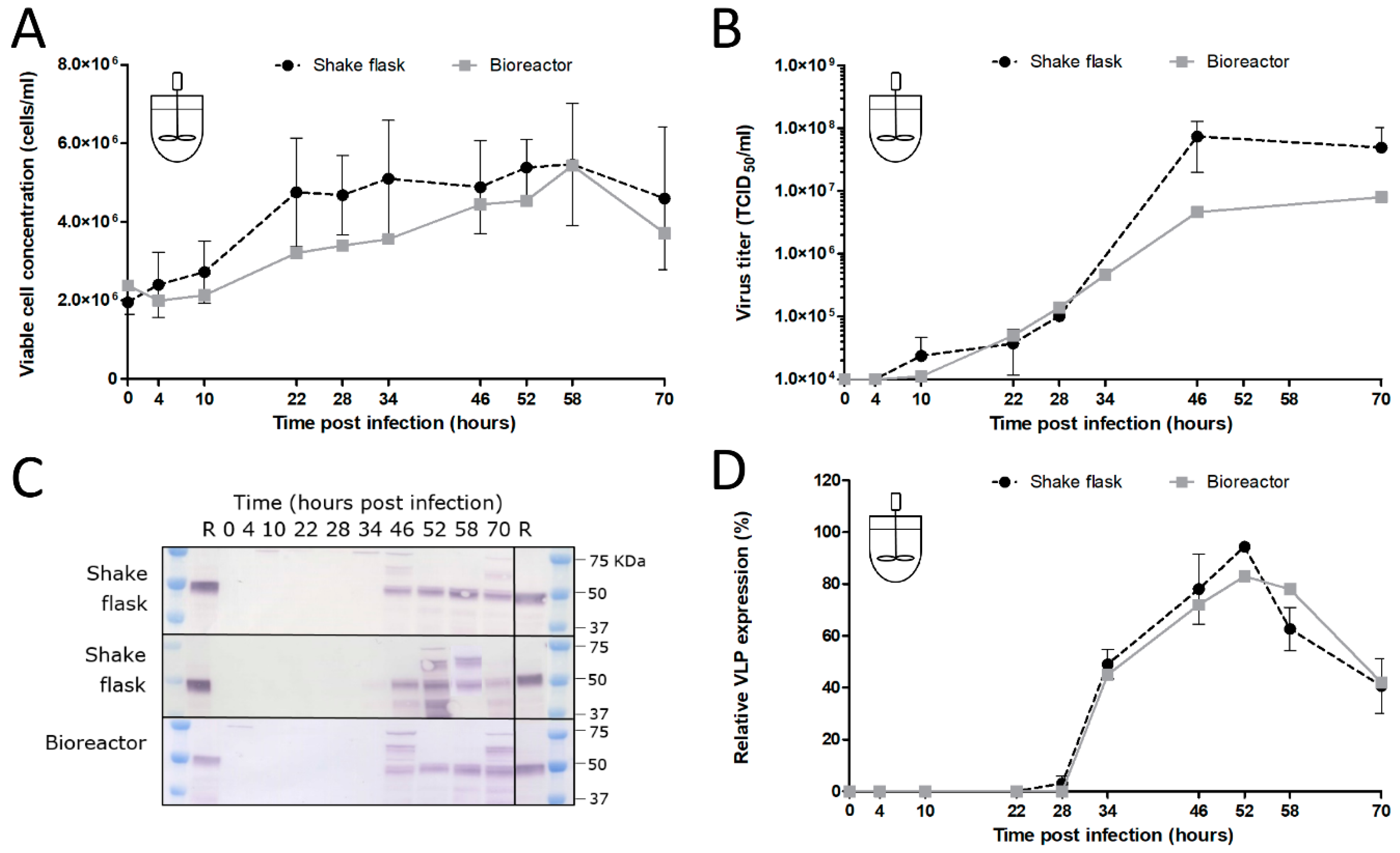
2.7. Shake Flask and Bioreactor Experiments
In the shake flask experiments, Sf9 suspension cells were cultured in Sf-900 II SFM medium (Gibco, Life Technologies, Carlsbad, CA, USA) in 125 mL flasks (Nalgene, Rochester, NY, USA) with a working volume of 25 mL. The culture conditions of the cells were identical to the culture conditions of the maintenance culture (27 °C, 100 rpm, inoculation at 0.5 × 106 cells/mL from an exponential phase cell culture at ~4 × 106 cells/mL). When the desired cell concentration was reached in the shake flasks, infection with the recombinant baculovirus was performed at a defined MOI. Samples were obtained at specific time points to analyze the infection process. A 1-litre DASGIP bioreactor (Eppendorf, Hamburg, Germany) was inoculated at 0.5 × 106 cells/mL by the Sf9 culture stock in the exponential growth phase. The bioreactors had a working volume of 500 mL and cells were maintained in Sf-900 II SFM medium (Gibco, Life Technologies, Carlsbad, CA, USA) at 27 °C and an agitation speed of 150 rpm. The DO (DO sensor, Broadley James, Irvine, CA, USA) was controlled at 30% air saturation by headspace aeration. The pH (pH sensor, Mettler Toledo, Tiel, the Netherlands) was retained at a value of 6.3 by automatic base and CO2 addition. Once the cells reached the desired CCI, infection with recombinant baculoviruses was performed at a defined MOI. Samples were obtained at several time points during the infection process.
4. Discussion
In this study, we scouted the AcMNPV genome for additional landing pads for the attTn7 transposition insertion site to present new options for the insertion of heterologous genes. These regions were initially selected based on data from a previous manuscript, which looked at direct insertion of recombinant protein coding regions into the baculovirus genome [
21]. We chose several of the most interesting regions from that work to examine in the context of the Tn7 transposition system. Each region had a unique expression capacity and is, therefore, useful for future experimentation. Most notably, BACe56 (DE37) consistently produced higher levels of protein expression than the control bacmid strain, with some proteins generating 1.4 times the original levels of recombinant protein. This suggests that some aspects of this location, whether due to the stability of the insertion or to some unknown transcriptional enhancer, lead to higher levels of protein production. While a 1.4 time increase in yield might appear to be minimal at a first glance, such an improvement in the production of proteins for therapeutic usage or as a vaccine product could carry significant reductions in cost of operations or the scale of production required.
Two of the other strains, DE38 (BAC51/52) and DE40 (v-Ubi/39k), with similar expression levels to the original polh locus, could potentially be used as alternative sites for the introduction of heterologous genes, or might prove useful as secondary insertion locations if the expression of multiple genes is desired. While the 2x reduced expression from the DE39 (gp37/DNApol) strain may seem to be a negative attribute of this location, in the case of slightly toxic proteins, or proteins which suffer from aggregation or solubility issues when overexpressed, the reduced expression level of DE39 might prove a good solution.
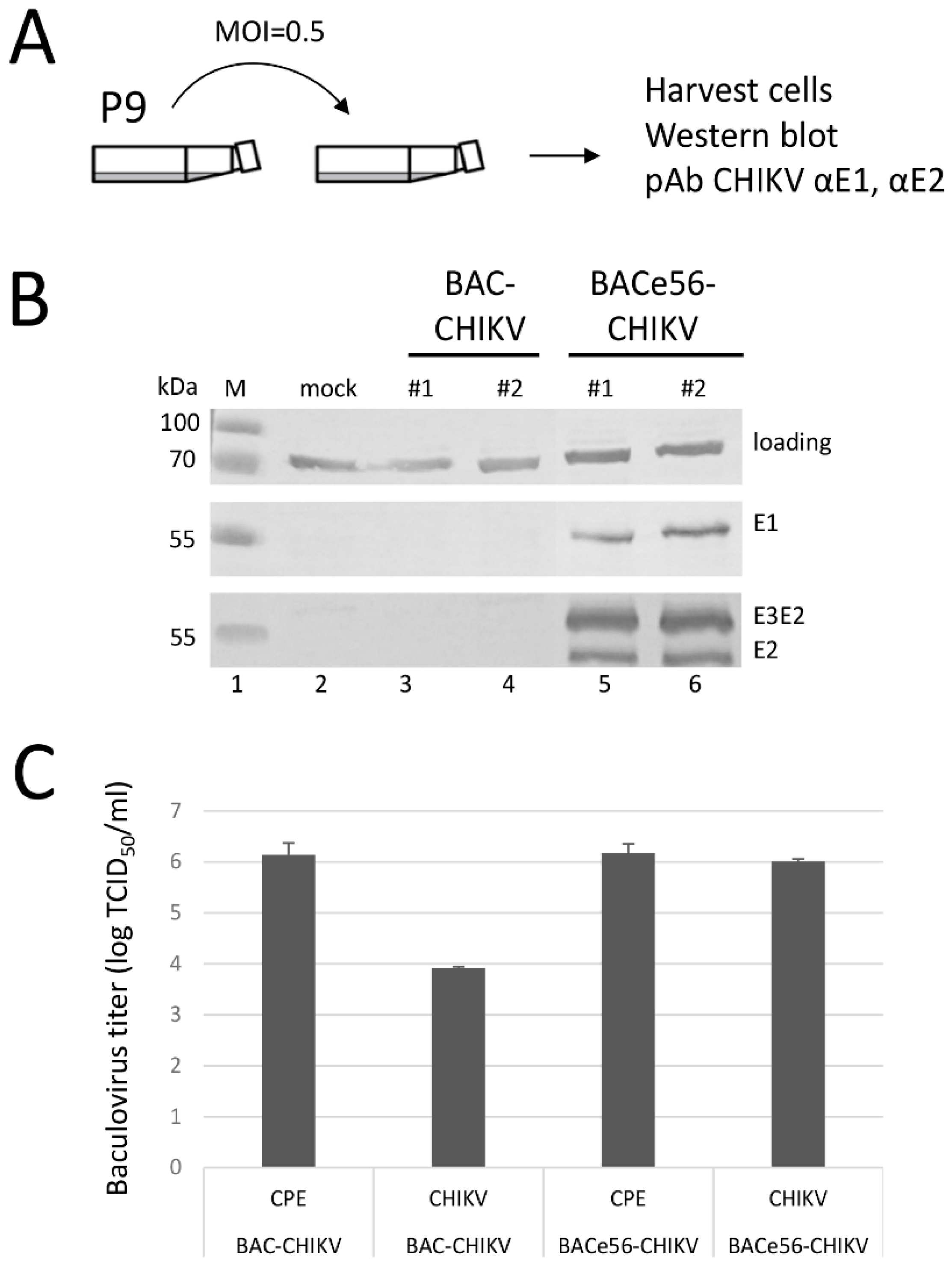
The serial undiluted passaging experiments very clearly showed that it was beneficial to separate the GOI from the unstable mini-F replicon in the bacmid. Based on the results, we could conclude that GOI expression from the odv-e56 (pif-5) locus resulted in the highest overall expression levels and increased stability upon serial passage compared to the original bacmid. The loss of GOI expression upon serial passage of original bacmid constructs was more rapid than what was seen in an earlier study [
19]. However, the size of the GOI in this study (CHIKV structural genes, 3.8 kbp) is much larger than the GFP gene (0.7 kbp), which increases the chances of deletion. Given the non-essential nature of the miniF replicon for baculovirus infection, the enhanced GOI stability in BACe56 vs. the original bacmid most likely relates to the relocation of the attTn7 site to a more stable location in the genome. In fact, odv-e56 is located in between baculovirus essential genes (ie-1 and ie-2) that cannot be deleted. Furthermore, instability of certain regions may correlate with a low density of baculovirus homologous repeat regions (hrs), which are dispersed throughout the baculovirus genome and are believed to act as origins of viral DNA replication (oris) [
29]. The odv-e56 gene is located quite close to hr1, which may enhance its stability in the genome and perhaps influences the transcriptional activity of nearby genes by functioning as an enhancer. This may explain why BACe56 gives slightly higher recombinant protein yields.
As the demand for the production of recombinant protein complexes increases, the four new bacmids generated in this study will be useful tools for optimizing expression and quality, while remaining the advantages of expression during the very late stage of the lytic cycle. Although these next-generation bacmids have been developed to be compatible with the Bac-to-Bac system, they can be used in combination with any Tn7-based baculovirus cloning method.
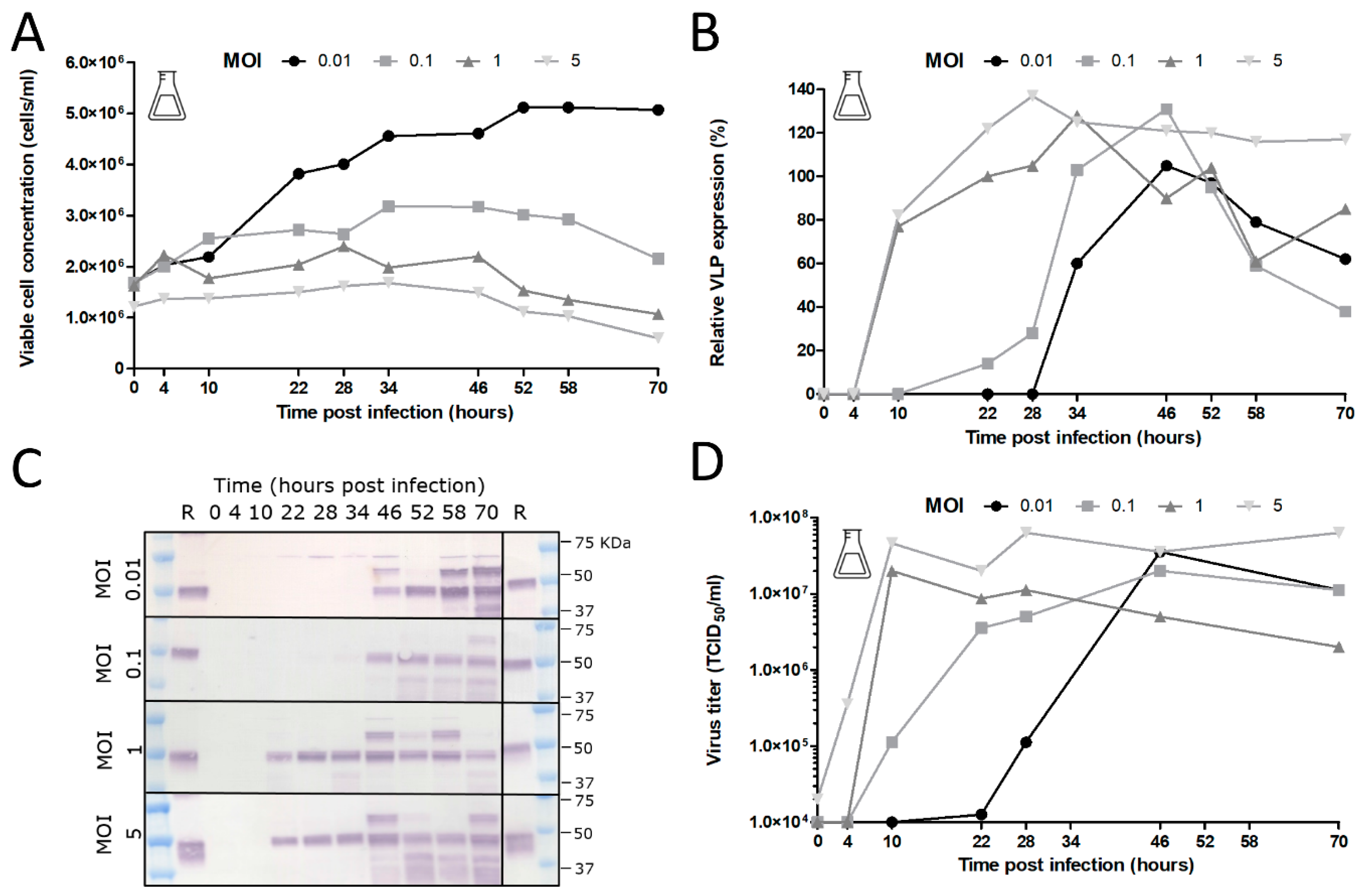
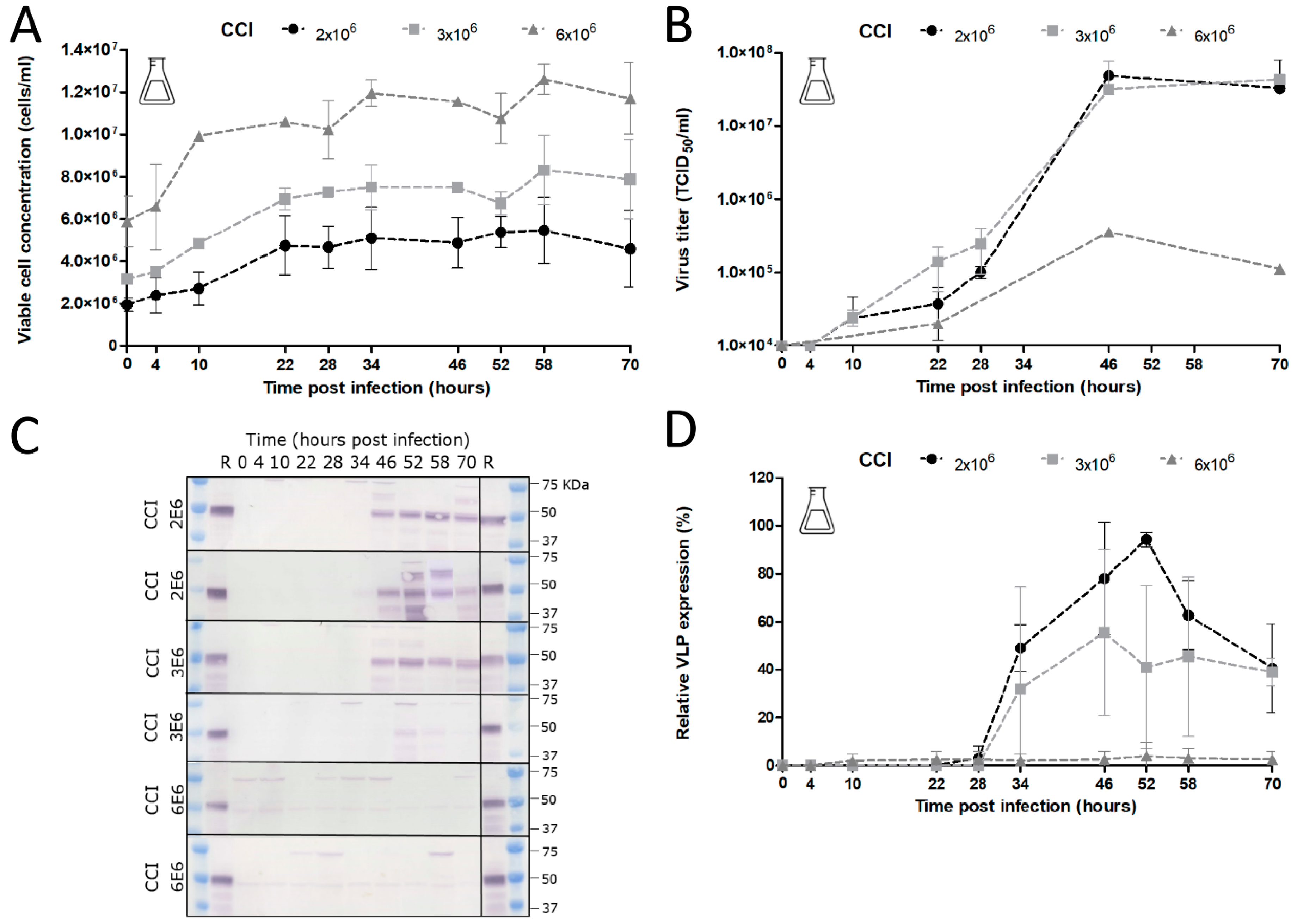
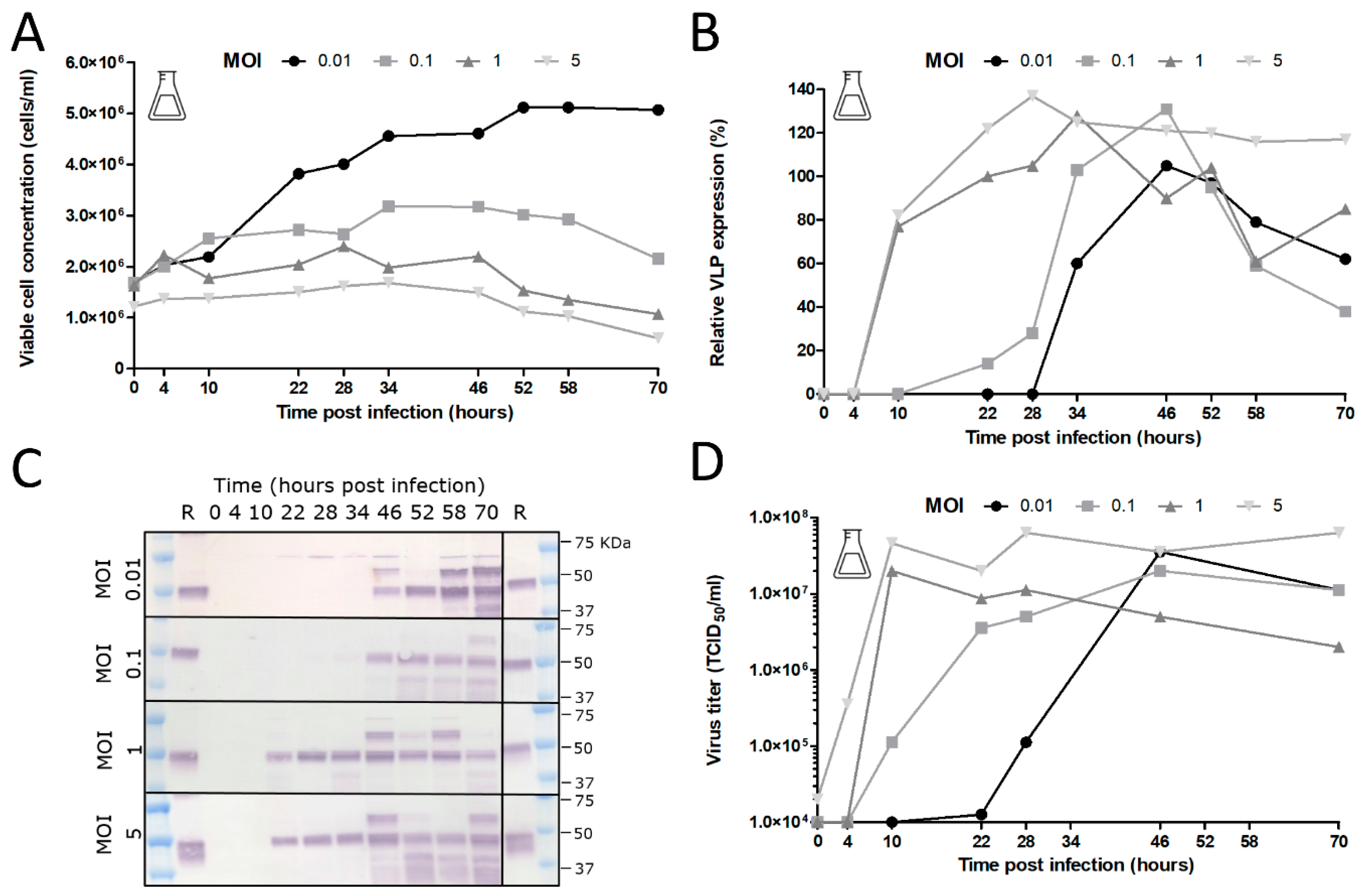
The added stability of GOI expression upon serial undiluted passage of BACe56 could have major impacts on its utility for large-scale protein production, which we examined in shake flask and bioreactor experiments. The interplay between MOI, CCI and time of harvest (TOH) of the new bacmid BACe56-expressing CHIKV VLPs in Sf9 suspension cells was investigated. Overall, the reported results showed a clear difference between infections at high MOIs (1 and 5) and low MOIs (0.01 and 0.1), which is in line with other studies [
30,
31,
32,
33]. Infection of the insect cells at lower MOIs lead to 1–2 infection cycles in the culture and to the proliferation of the uninfected cells and thus higher maximum cell concentrations. Infection at an MOI of five immediately inhibited cell growth because all cells were infected simultaneously. The differences in cell growth, viral infection process and VLP expression observed between infections at diverse MOIs highlights the importance of this infection parameter in the establishment of a production process for recombinant baculovirus-expressed VLPs. Infection at an MOI of 0.01 is preferred for further process developments, since comparable product yields were reached because higher MOIs and smaller quantities of virus are required, which may obliviate the use of additional bioreactors for virus production.
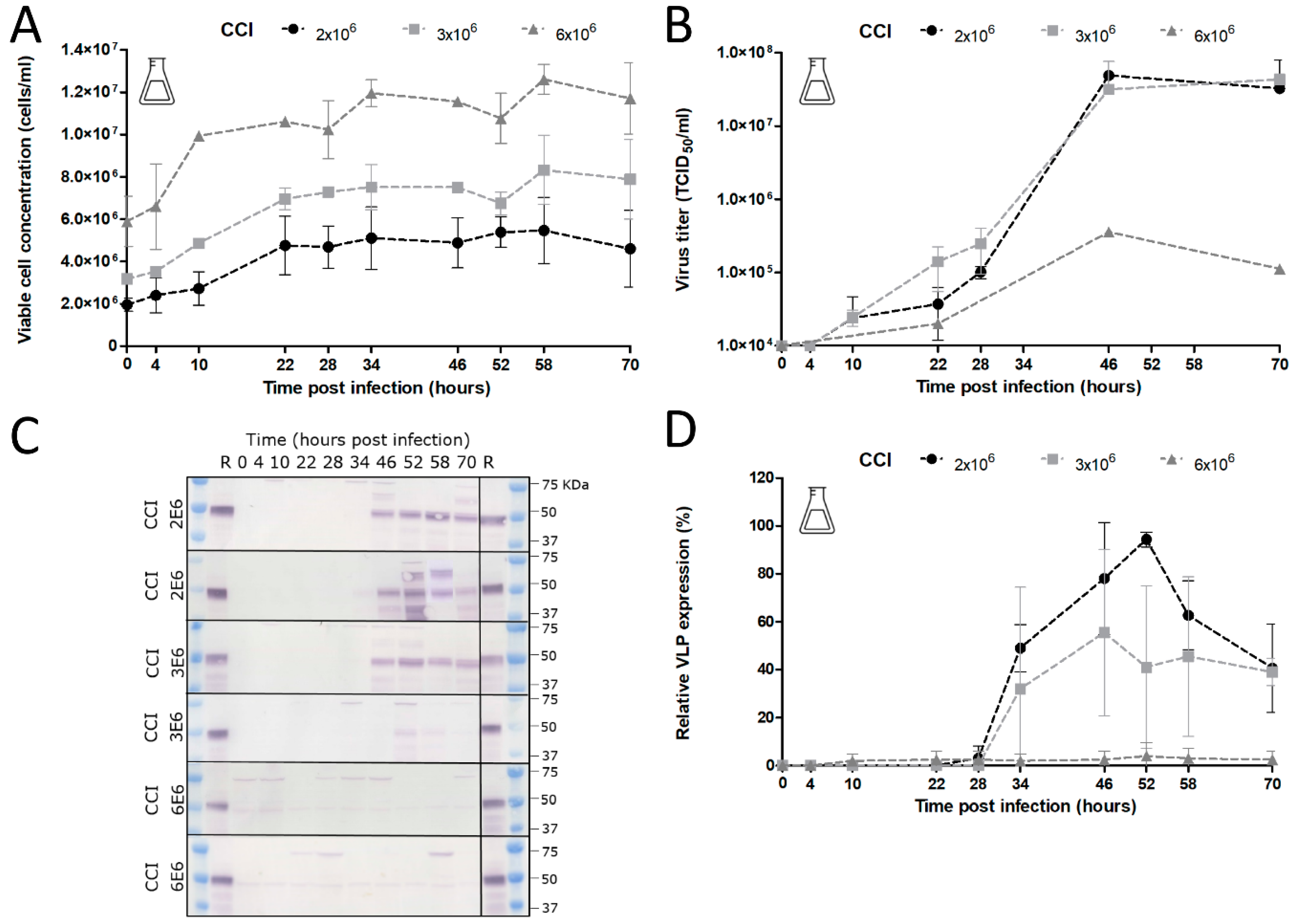
The choice of CCI is also very important since it can cause significant alterations in product yield, especially when low-MOI infection is applied. Baculovirus infection above a certain optimum CCI leads to an ineffective infection with poor baculovirus titers and low product concentrations [
32,
34,
35]. Indeed, we observed unsuccessful infection at a CCI above 4 × 10
6 cells/mL with no detectable CHIKV VLP yields. The optimum VLP production was achieved at a CCI of 2 × 10
6 cells/mL, whereas infections at CCIs higher than 3 × 10
6 cells/mL resulted in a reduction in baculovirus infection and VLP production. With regard to specific protein yields, we generated overall CHIKV VLP yields between 2–4 mg/L. In similar systems, the yields varied from 0.2 mg/L for SARS coronavirus VLPs [
36] to 662 mg/L for rotavirus VLPs [
37]. Most likely, further optimization of CHIKV VLP yields can be accomplished since CHIKV VLPs up to 40 mg/L were produced in adherent Sf21 cells [
22], while 28 mg/L CHIKV VLPs were produced in in Sf21-derived suspension Sf-basic cells [
38]. We conclude that BACe56 is a stable vector with improved expression characteristics that is suitable for the production of complex VLP vaccines in insect cell bioreactors.


 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}