
Multiple Introductions Followed by Ongoing Community Spread of SARS-CoV-2 at One of the Largest Metropolitan Areas of Northeast Brazil

 , , , , , , , , , , add
Show full author list
, , , , , , , , , , add
Show full author list
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Samples and SARS-CoV-2 Molecular Detection
2.2. Genomic Sequencing
2.3. Viral Strain Isolation and Resequencing
2.4. Genome Assembly and Annotation
2.5. Evolutionary Analysis
2.6. Single Nucleotide Polymorphism Analysis
2.7. Epidemiological Data
3. Results and Discussion
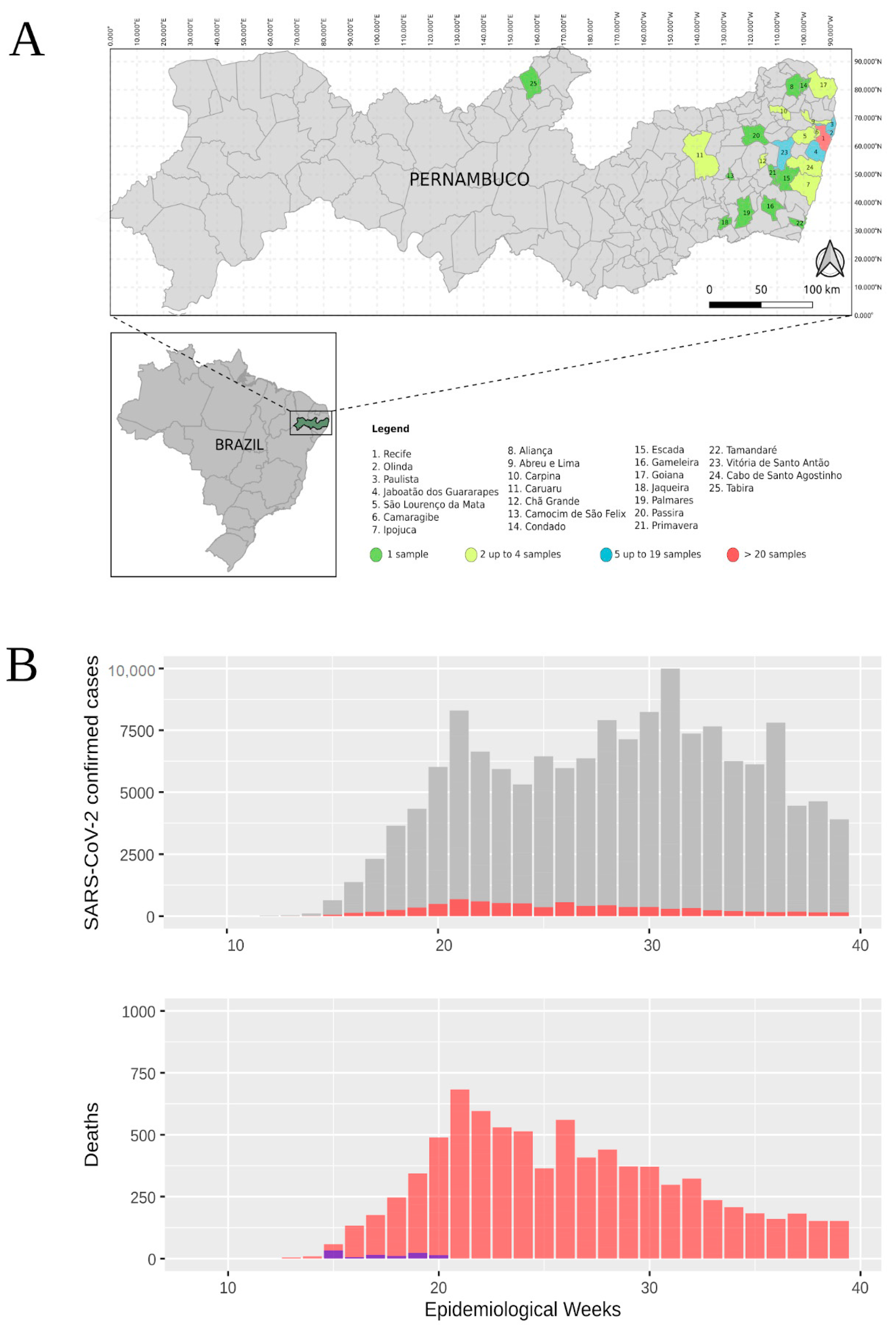
3.1. Epidemiological Data from Pernambuco State
3.2. Genome Sequencing and Variability
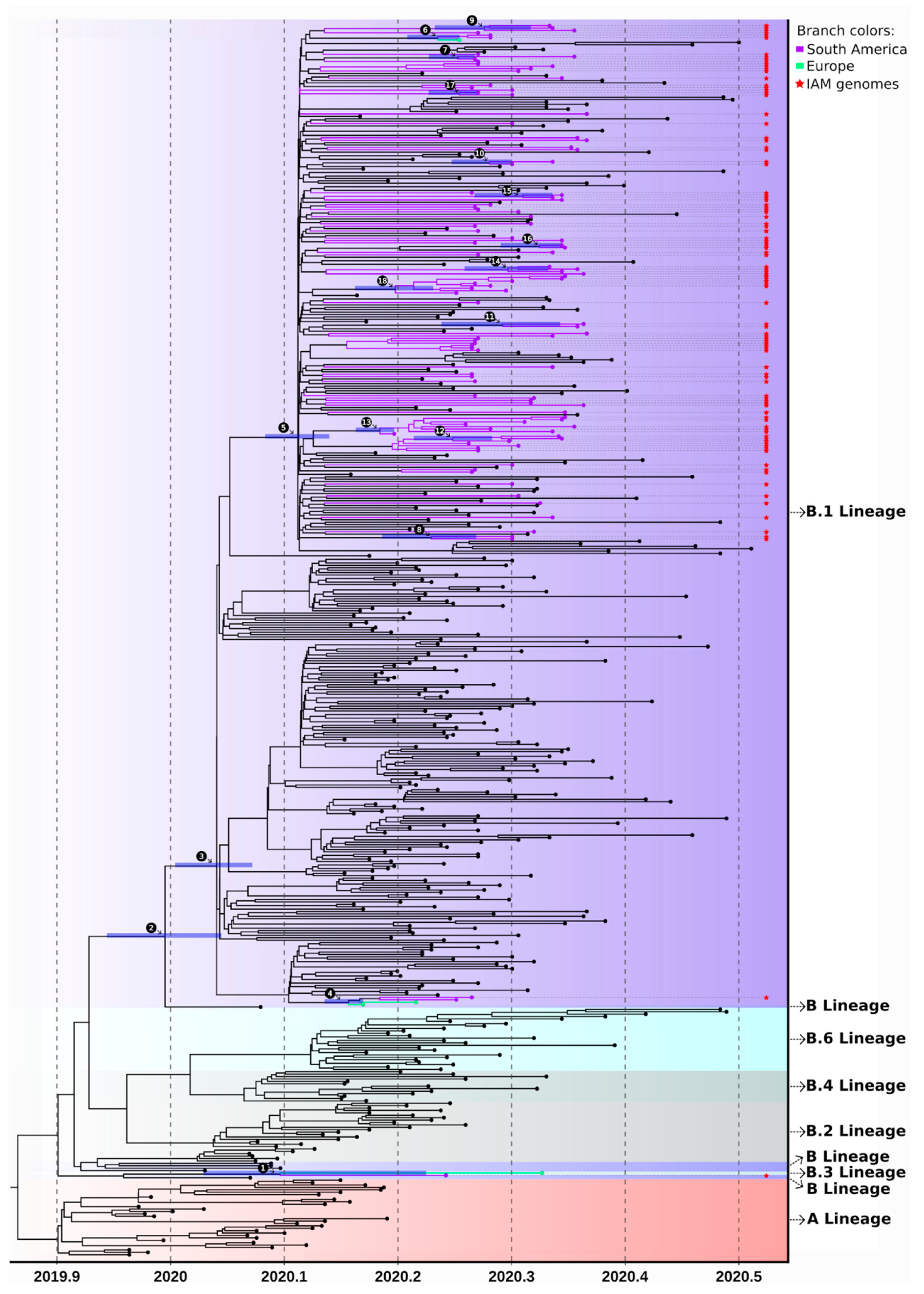
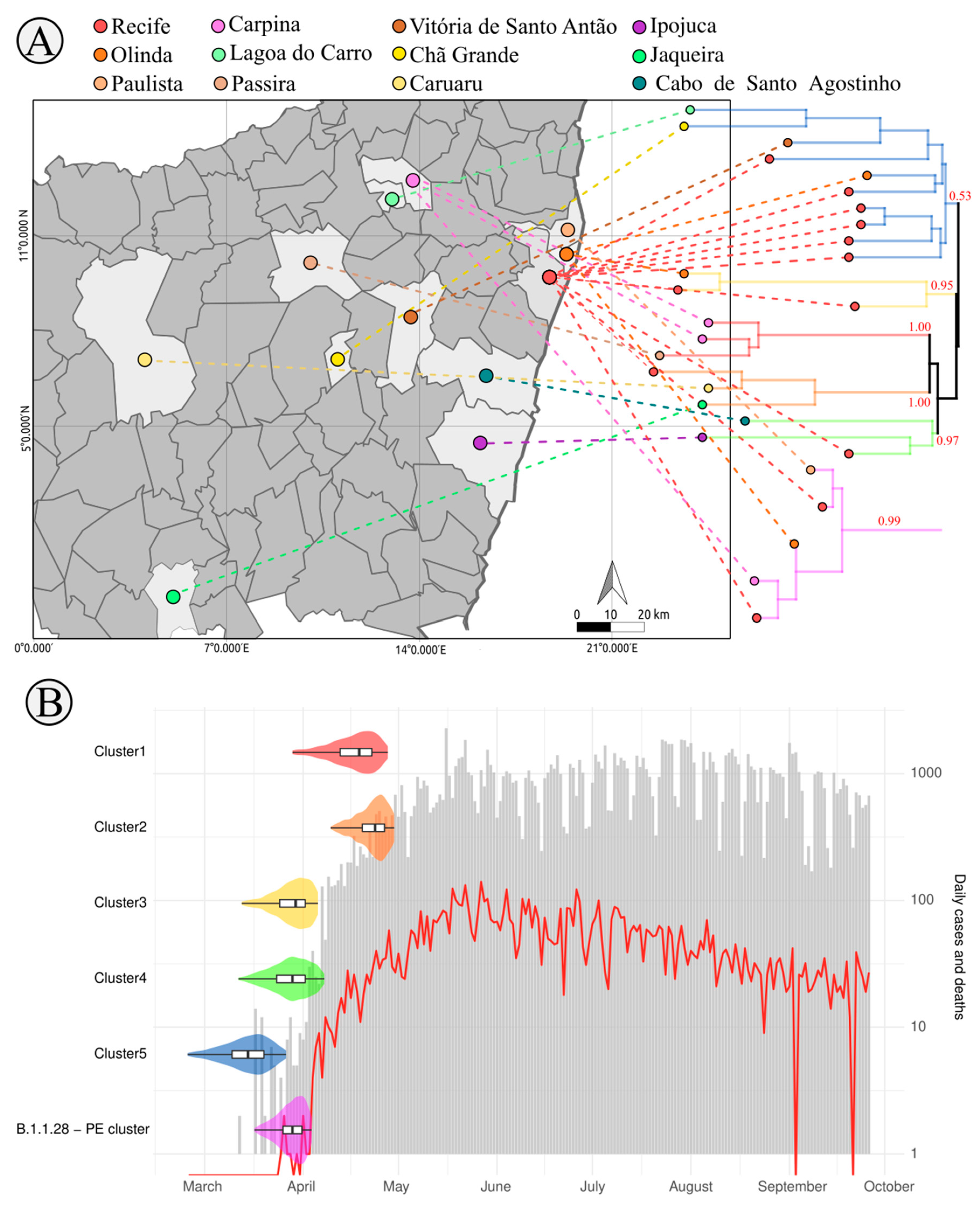
3.3. SARS-CoV-2 Lineages in Pernambuco
4. Conclusions
Data availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yang, X.; Yu, Y.; Xu, J.; Shu, H.; Xia, J.; Liu, H.; Wu, Y.; Zhang, L.; Yu, Z.; Fang, M.; et al. Clinical course and outcomes of critically ill patients with SARS-CoV-2 pneumonia in Wuhan, China: A single-centered, retrospective, observational study. Lancet Respir. Med. 2020, 8, 475481. [Google Scholar] [CrossRef]
- Rabi, F.A.; Al Zoubi, M.S.; Kasasbeh, G.A.; Salameh, D.M.; Al-Nasser, A.D. SARS-CoV-2 and Coronavirus Disease 2019: What We Know So Far. Pathogens 2020, 9, 231. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270273. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Lau, E.H.Y.; Wu, P.; Deng, X.; Wang, J.; Hao, X.; Lau, Y.C.; Wong, J.Y.; Guan, Y.; Tan, X.; et al. Temporal dynamics in viral shedding and transmissibility of COVID-19. Nat. Med. 2020, 26, 672675. [Google Scholar] [CrossRef] [PubMed]
- Tuite, A.R.; Ng, V.; Rees, E.; Fisman, D. Estimation of COVID-19 outbreak size in Italy. Lan. Infec. Deseases 2020, 20, 537. [Google Scholar] [CrossRef]
- Licastro, D.; Rajasekharan, S.; Dal Monego, S.; Segat, L.; DAgaro, P.; Marcello, A. Isolation and Full-Length Genome Characterization of SARS-CoV-2 from COVID-19 Cases in Northern Italy. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Gonzalez-Reiche, A.S.; Hernandez, M.M.; Sullivan, M.J.; Ciferri, B.; Alshammary, H.; Obla, A.; Fabre, S.; Kleiner, G.; Polanco, J.; Khan, Z.; et al. Introductions and early spread of SARS-CoV-2 in the New York City area. Science 2020, eabc1917. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533534. [Google Scholar] [CrossRef]
- de Souza, W.M.; Buss, L.F.; da Silva Candido, D.; Carrera, J.-P.; Li, S.; Zarebski, A.E.; Pereira, R.H.M.; Prete, C.A.; de Souza-Santos, A.A.; Parag, K.V.; et al. Epidemiological and clinical characteristics of the COVID-19 epidemic in Brazil. Nat. Hum. Behav. 2020, 4, 856–865. [Google Scholar] [CrossRef]
- do Nascimento, V.A.; Corado, A.L.G.; do Nascimento, F.O.; da Costa, K.A.; Duarte, D.C.G.; de Jesus, M.S.; Luz, S.L.B.; Gonalves, L.M.F.; da Costa, C.F.; Delatorre, E. Genomic and phylogenetic characterization of an imported case of SARS-CoV-2 in Amazonas State, Brazil. Memorias Instituto Oswaldo Cruz 2017, 115, e200310. [Google Scholar] [CrossRef]
- Xavier, J.; Giovanetti, M.; Adelino, T.; Fonseca, V.; da Costa, A.V.B.; Ribeiro, A.A.; Felicio, K.N.; Duarte, C.G.; Silva, M.V.F.; Lima, M.T.; et al. The ongoing COVID-19 epidemic in Minas Gerais, Brazil: Insights from epidemiological data and SARS-CoV-2 whole genome sequencing. Emerg. Microbes Infect. 2020, 9, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Resende, P.C.; Motta, F.C.; Roy, S.; Appolinario, L.; Fabri, A.; Xavier, J.; Harris, K.; Matos, A.R.; Caetano, B.; Orgeswalska, M.; et al. SARS-CoV-2 genomes recovered by long amplicon tiling multiplex approach using nanopore sequencing and applicable to other sequencing platforms. BioRxiv 2020. [Google Scholar] [CrossRef]
- Candido, D.S.; Claro, I.M.; de Jesus, J.G.; Souza, W.M.; Moreira, F.R.R.; Dellicour, S.; Mellan, T.A.; du Plessis, L.; Pereira, R.H.M.; Sales, F.C.S.; et al. Evolution and epidemic spread of SARS-CoV-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef]
- Grubaugh, N.D.; Ladner, J.T.; Lemey, P.; Pybus, O.G.; Rambaut, A.; Holmes, E.C.; Andersen, K.G. Tracking virus outbreaks in the twenty-first century. Nat. Microbiol. 2018, 4, 1019. [Google Scholar] [CrossRef] [PubMed]
- WHO Laboratory Testing for 2019 Novel Coronavirus (2019-nCoV) in Suspected Human Cases; World Health Organ: Geneva, Switzerland, 2020.
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brnink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357359. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R. BEDTools: The Swiss-Army Tool for Genome Feature Analysis. Curr. Protoc. Bioinforma. 2014, 47. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 20782079. [Google Scholar] [CrossRef]
- Danecek, P.; Auton, A.; Abecasis, G.; Albers, C.A.; Banks, E.; DePristo, M.A.; Handsaker, R.E.; Lunter, G.; Marth, G.T.; Sherry, S.T.; et al. 1000 Genomes Project Analysis Group The variant call format and VCFtools. Bioinformatics 2011, 27, 21562158. [Google Scholar] [CrossRef]
- Gel, B.; Serra, E. karyoploteR: An R/Bioconductor package to plot customizable genomes displaying arbitrary data. Bioinformatics 2017, 33, 30883090. [Google Scholar] [CrossRef]
- Dezordi, F.Z. SARS-COV-TOOLS. Available online: https://github.com/dezordi/SARS-CoV-2_tools (accessed on 3 June 2020).
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772780. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Holmes, E.C.; Toole, O.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A dynamic nomenclature proposal for SARS-CoV-2 lineages to assist genomic epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Godzik, A. Cd-hit: A fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 2006, 22, 16581659. [Google Scholar] [CrossRef] [PubMed]
- Larsson, A. AliView: A fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 2014, 30. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268274. [Google Scholar] [CrossRef]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the Ultrafast Bootstrap Approximation. Mol. Biol. Evol. 2018, 35, 518522. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587589. [Google Scholar] [CrossRef]
- Lemoine, F.; Entfellner, J.-B.D.; Wilkinson, E.; Correia, D.; Felipe, M.D.; Oliveira, T.D.; Gascuel, O. Renewing Felsensteins phylogenetic bootstrap in the era of big data. Nature 2018, 556, 452456. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v4: Recent updates and new developments. Nucleic Acids Res. 2019, 47, W256W259. [Google Scholar] [CrossRef]
- Rambaut, A.; Lam, T.T.; Carvalho, L.M.; Pybus, O.G. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [PubMed]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal signal and the phylodynamic threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef] [PubMed]
- Page, A.J.; Taylor, B.; Delaney, A.J.; Soares, J.; Seemann, T.; Keane, J.A.; Harris, S.R. SNP-sites: Rapid efficient extraction of SNPs from multi-FASTA alignments. Microb. Genom. 2016, 2, e000056. [Google Scholar] [CrossRef] [PubMed]
- Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 2011, 27, 29872993. [Google Scholar] [CrossRef] [PubMed]
- BAM-READCOUNT. Available online: https://github.com/genome/bam-readcount (accessed on 3 June 2020).
- Liu, Z.; Zheng, H.; Lin, H.; Li, M.; Yuan, R.; Peng, J.; Xiong, Q.; Sun, J.; Li, B.; Wu, J.; et al. Identification of common deletions in the spike protein of SARS-CoV-2. J. Virol. 2020. [Google Scholar] [CrossRef]
- de Magalhes, J.J.F.; Mendes, R.P.G.; da Silva, C.T.A.; da Silva, S.J.R.; Guarines, K.M.; Pena, L. Epidemiological and clinical characteristics of the first 557 successive patients with COVID-19 in Pernambuco state, Northeast Brazil. Travel Med. Infect. Dis. 2020, 38, 101884. [Google Scholar] [CrossRef] [PubMed]
- Fung, T.S.; Liu, D.X. Human Coronavirus: Host-Pathogen Interaction. Annu. Rev. Microbiol. 2019, 73, 529557. [Google Scholar] [CrossRef]
- Li, Q.; Wu, J.; Nie, J.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; Zhang, Q.; Liu, H.; Nie, L.; et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Hengartner, N.; Giorgi, E.E.; Bhattacharya, T.; Foley, B.; et al. Tracking changes in SARS-CoV-2 Spike: Evidence that D614G increases infectivity of the COVID-19 virus. Cell 2020, 182, 812–827. [Google Scholar] [CrossRef]
- Becerra-Flores, M.; Cardozo, T. SARS-CoV-2 viral spike G614 mutation exhibits higher case fatality rate. Int. J. Clin. Pract. 2020, e13525. [Google Scholar] [CrossRef]
- Yurkovetskiy, L.; Wang, X.; Pascal, K.E.; Tomkins-Tinch, C.; Nyalile, T.P.; Wang, Y.; Baum, A.; Diehl, W.E.; Dauphin, A.; Carbone, C.; et al. Structural and Functional Analysis of the D614G SARS-CoV-2 Spike Protein Variant. Cell 2020. [Google Scholar] [CrossRef] [PubMed]
- Grubaugh, N.D.; Hanage, W.P.; Rasmussen, A.L. Making sense of mutation: What D614G means for the COVID-19 pandemic remains unclear. Cell 2020, 182, 794–795. [Google Scholar] [CrossRef] [PubMed]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Stefanelli, P.; Faggioni, G.; Lo Presti, A.; Fiore, S.; Marchi, A.; Benedetti, E.; Fabiani, C.; Anselmo, A.; Ciammaruconi, A.; Fortunato, A.; et al. Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: Additional clues on multiple introductions and further circulation in Europe. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Angeletti, S.; Benvenuto, D.; Ciccozzi, M. A doubt of multiple introduction of SARS-CoV-2 in Italy: A preliminary overview. J. Med. Virol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.R.; Graham, R.L.; Donaldson, E.F.; Eckerle, L.D.; Baric, R.S. Coronaviruses: An RNA proofreading machine regulates replication fidelity and diversity. RNA Biol. 2011, 8, 270279. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paiva, M.H.S.; Guedes, D.R.D.; Docena, C.; Bezerra, M.F.; Dezordi, F.Z.; Machado, L.C.; Krokovsky, L.; Helvecio, E.; da Silva, A.F.; Vasconcelos, L.R.S.; et al. Multiple Introductions Followed by Ongoing Community Spread of SARS-CoV-2 at One of the Largest Metropolitan Areas of Northeast Brazil. Viruses 2020, 12, 1414. https://doi.org/10.3390/v12121414
Paiva MHS, Guedes DRD, Docena C, Bezerra MF, Dezordi FZ, Machado LC, Krokovsky L, Helvecio E, da Silva AF, Vasconcelos LRS, et al. Multiple Introductions Followed by Ongoing Community Spread of SARS-CoV-2 at One of the Largest Metropolitan Areas of Northeast Brazil. Viruses. 2020; 12(12):1414. https://doi.org/10.3390/v12121414
Chicago/Turabian StylePaiva, Marcelo Henrique Santos, Duschinka Ribeiro Duarte Guedes, Cássia Docena, Matheus Filgueira Bezerra, Filipe Zimmer Dezordi, Laís Ceschini Machado, Larissa Krokovsky, Elisama Helvecio, Alexandre Freitas da Silva, Luydson Richardson Silva Vasconcelos, and et al. 2020. "Multiple Introductions Followed by Ongoing Community Spread of SARS-CoV-2 at One of the Largest Metropolitan Areas of Northeast Brazil" Viruses 12, no. 12: 1414. https://doi.org/10.3390/v12121414
APA StylePaiva, M. H. S., Guedes, D. R. D., Docena, C., Bezerra, M. F., Dezordi, F. Z., Machado, L. C., Krokovsky, L., Helvecio, E., da Silva, A. F., Vasconcelos, L. R. S., Rezende, A. M., da Silva, S. J. R., Sales, K. G. d. S., de Sá, B. S. L. F., da Cruz, D. L., Cavalcanti, C. E., Neto, A. d. M., da Silva, C. T. A., Mendes, R. P. G., ... Wallau, G. L. (2020). Multiple Introductions Followed by Ongoing Community Spread of SARS-CoV-2 at One of the Largest Metropolitan Areas of Northeast Brazil. Viruses, 12(12), 1414. https://doi.org/10.3390/v12121414