
The Crosstalk of Epigenetics and Metabolism in Herpesvirus Infection



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Aerobic Glycolysis or Warburg Effect
3. Fatty Acid Synthesis
4. S-Adenosylmethionine (SAM)
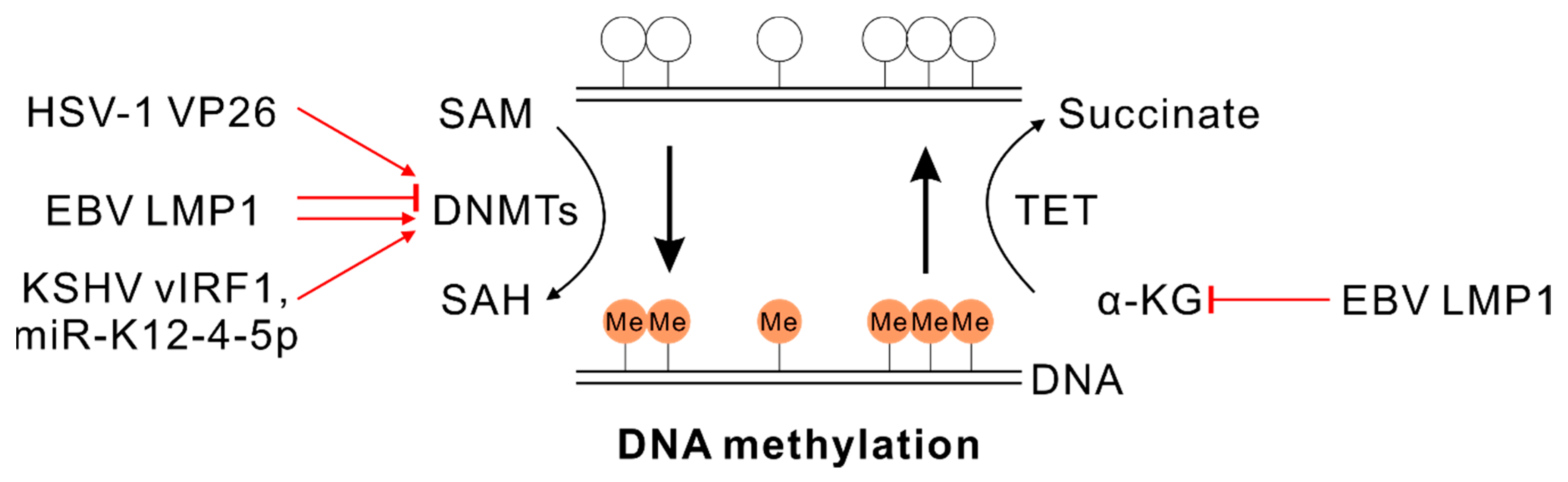
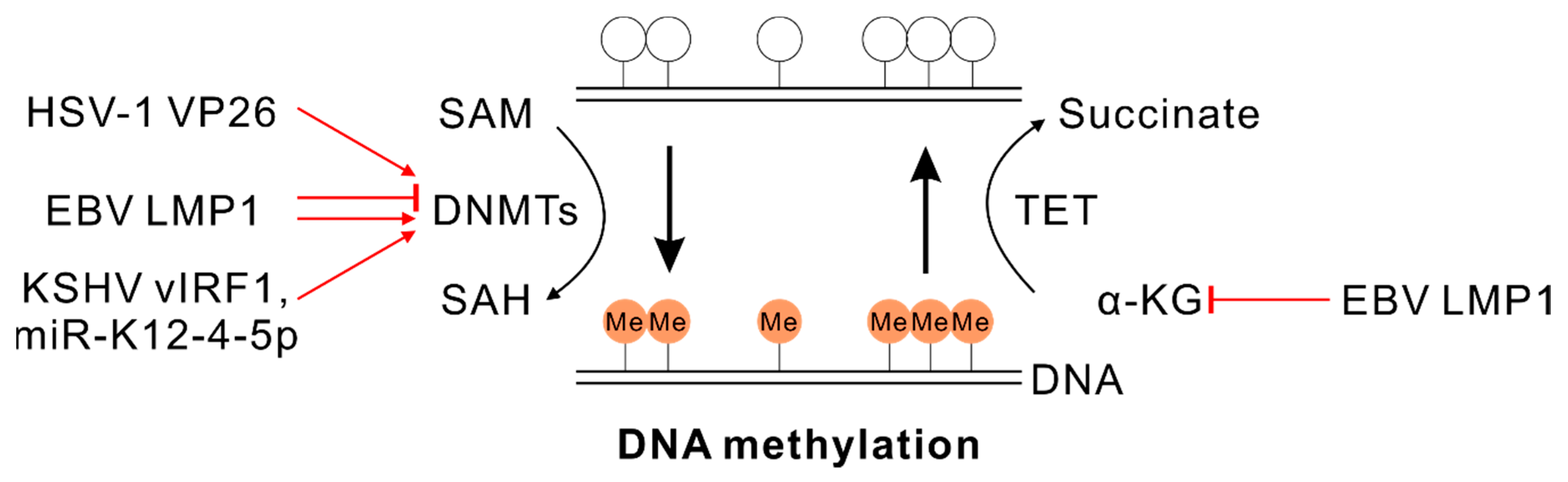
4.1. SAM and DNA Methylation
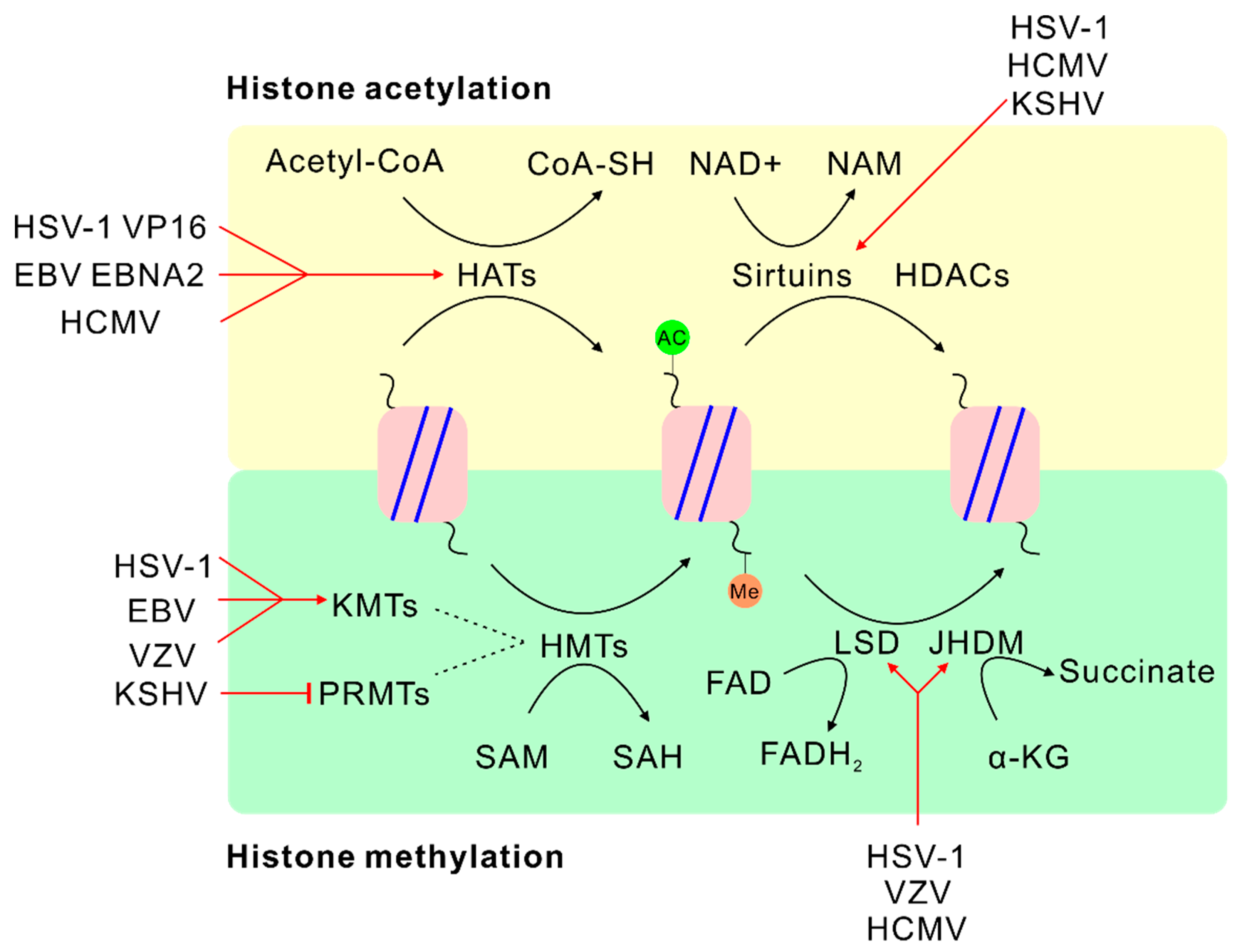
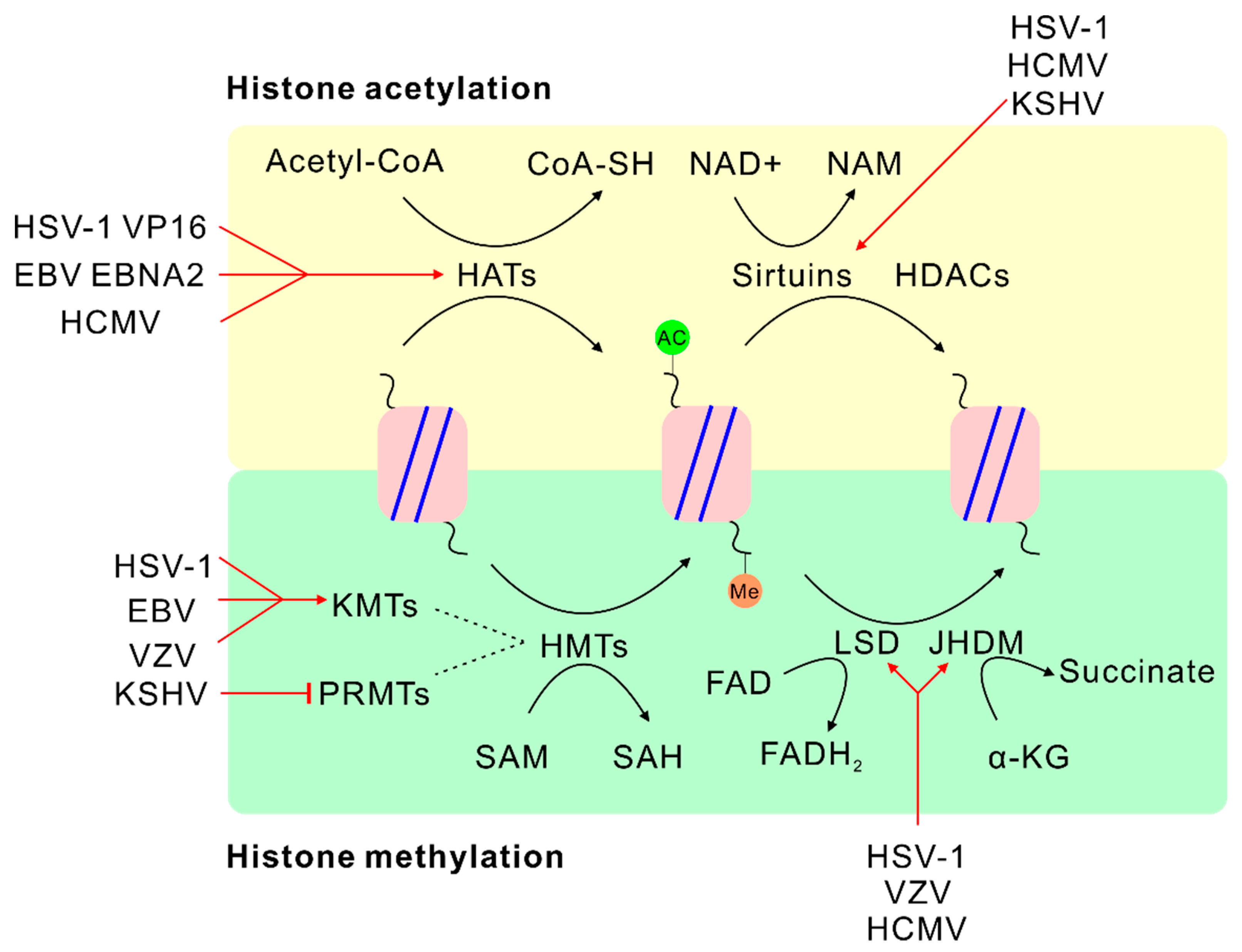
4.2. SAM and Histone Methylation
5. α-Ketoglutarate (α-KG) and Flavin Adenine Dinucleotide (FAD)
5.1. α-KG and DNA Demethylation
5.2. α-KG or FAD and Histone Demethylation
6. Acetyl-Coenzyme A (Acetyl-CoA)
7. Nicotinamide Adenine Dinucleotide (NAD+)
8. Glutaminolysis
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fields, B.N.; Knipe, D.M.; Howley, P.M. Fields Virology, 6th ed.; Wolters Kluwer Health/Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Cohen, J.I. Herpesvirus latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef]
- Lieberman, P.M. Epigenetics and Genetics of Viral Latency. Cell Host Microbe 2016, 19, 619–628. [Google Scholar] [CrossRef] [Green Version]
- Kristie, T.M. Dynamic modulation of HSV chromatin drives initiation of infection and provides targets for epigenetic therapies. Virology 2015, 479–480, 555–561. [Google Scholar] [CrossRef] [Green Version]
- Cliffe, A.R.; Coen, D.M.; Knipe, D.M. Kinetics of facultative heterochromatin and polycomb group protein association with the herpes simplex viral genome during establishment of latent infection. mBio 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Chung, C.; Sweha, S.R.; Pratt, D.; Tamrazi, B.; Panwalkar, P.; Banda, A.; Bayliss, J.; Hawes, D.; Yang, F.; Lee, H.J.; et al. Integrated Metabolic and Epigenomic Reprograming by H3K27M Mutations in Diffuse Intrinsic Pontine Gliomas. Cancer Cell 2020. [Google Scholar] [CrossRef]
- Dai, Z.; Ramesh, V.; Locasale, J.W. The evolving metabolic landscape of chromatin biology and epigenetics. Nat. Rev. Genet. 2020. [Google Scholar] [CrossRef]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [Green Version]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, X.; Hong, L.; Cheng, C.; Li, N.; Zhao, X.; Shi, F.; Liu, J.; Fan, J.; Zhou, J.; Bode, A.M.; et al. DNMT1 mediates metabolic reprogramming induced by Epstein-Barr virus latent membrane protein 1 and reversed by grifolin in nasopharyngeal carcinoma. Cell Death Dis. 2018, 9, 619. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Hu, Z.Y.; Dong, X.; Tan, Z.; Li, W.; Tang, M.; Chen, L.; Yang, L.; Tao, Y.; Jiang, Y.; et al. Targeting Epstein-Barr virus oncoprotein LMP1-mediated glycolysis sensitizes nasopharyngeal carcinoma to radiation therapy. Oncogene 2014, 33, 4568–4578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hulse, M.; Caruso, L.B.; Madzo, J.; Tan, Y.; Johnson, S.; Tempera, I. Poly(ADP-ribose) polymerase 1 is necessary for coactivating hypoxia-inducible factor-1-dependent gene expression by Epstein-Barr virus latent membrane protein 1. PLoS Pathog. 2018, 14, e1007394. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006, 2, e132. [Google Scholar] [CrossRef] [Green Version]
- DeVito, S.R.; Ortiz-Riano, E.; Martinez-Sobrido, L.; Munger, J. Cytomegalovirus-mediated activation of pyrimidine biosynthesis drives UDP-sugar synthesis to support viral protein glycosylation. Proc. Natl. Acad. Sci. USA 2014, 111, 18019–18024. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landini, M.P. Early enhanced glucose uptake in human cytomegalovirus-infected cells. J. Gen. Virol. 1984, 65, 1229–1232. [Google Scholar] [CrossRef]
- Yu, Y.; Maguire, T.G.; Alwine, J.C. Human cytomegalovirus activates glucose transporter 4 expression to increase glucose uptake during infection. J. Virol. 2011, 85, 1573–1580. [Google Scholar] [CrossRef] [Green Version]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar] [CrossRef] [Green Version]
- McArdle, J.; Moorman, N.J.; Munger, J. HCMV targets the metabolic stress response through activation of AMPK whose activity is important for viral replication. PLoS Pathog. 2012, 8, e1002502. [Google Scholar] [CrossRef] [Green Version]
- McGee, S.L.; van Denderen, B.J.; Howlett, K.F.; Mollica, J.; Schertzer, J.D.; Kemp, B.E.; Hargreaves, M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes 2008, 57, 860–867. [Google Scholar] [CrossRef]
- McArdle, J.; Schafer, X.L.; Munger, J. Inhibition of calmodulin-dependent kinase kinase blocks human cytomegalovirus-induced glycolytic activation and severely attenuates production of viral progeny. J. Virol. 2011, 85, 705–714. [Google Scholar] [CrossRef] [Green Version]
- Delgado, T.; Carroll, P.A.; Punjabi, A.S.; Margineantu, D.; Hockenbery, D.M.; Lagunoff, M. Induction of the Warburg effect by Kaposi’s sarcoma herpesvirus is required for the maintenance of latently infected endothelial cells. Proc. Natl. Acad. Sci. USA 2010, 107, 10696–10701. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.L.; Pulliam, T.H.; Dimaio, T.A.; Thalhofer, A.B.; Delgado, T.; Lagunoff, M. Glycolysis, Glutaminolysis, and Fatty Acid Synthesis Are Required for Distinct Stages of Kaposi‘s Sarcoma-Associated Herpesvirus Lytic Replication. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.K.; Lang, F.; Pei, Y.; Jha, H.C.; Robertson, E.S. Metabolic reprogramming of Kaposi‘s sarcoma associated herpes virus infected B-cells in hypoxia. PLoS Pathog. 2018, 14, e1007062. [Google Scholar] [CrossRef] [Green Version]
- Yogev, O.; Lagos, D.; Enver, T.; Boshoff, C. Kaposi‘s sarcoma herpesvirus microRNAs induce metabolic transformation of infected cells. PLoS Pathog. 2014, 10, e1004400. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.M.; Tymoczko, J.L.; Stryer, L. Fatty Acids Are Synthesized and Degraded by Different Pathways. In Biochemistry, 5th ed.; W H Freeman: New York, NY, USA, 2002. [Google Scholar]
- Sanchez, E.L.; Lagunoff, M. Viral activation of cellular metabolism. Virology 2015, 479–480, 609–618. [Google Scholar] [CrossRef] [Green Version]
- Currie, E.; Schulze, A.; Zechner, R.; Walther, T.C.; Farese, R.V., Jr. Cellular fatty acid metabolism and cancer. Cell Metab. 2013, 18, 153–161. [Google Scholar] [CrossRef] [Green Version]
- Zaidi, N.; Swinnen, J.V.; Smans, K. ATP-citrate lyase: A key player in cancer metabolism. Cancer Res. 2012, 72, 3709–3714. [Google Scholar] [CrossRef] [Green Version]
- Brownsey, R.W.; Boone, A.N.; Elliott, J.E.; Kulpa, J.E.; Lee, W.M. Regulation of acetyl-CoA carboxylase. Biochem. Soc. Trans. 2006, 34, 223–227. [Google Scholar] [CrossRef]
- Jakobsson, A.; Westerberg, R.; Jacobsson, A. Fatty acid elongases in mammals: Their regulation and roles in metabolism. Prog. Lipid Res. 2006, 45, 237–249. [Google Scholar] [CrossRef]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Webster-Cyriaque, J.; Tomlinson, C.C.; Yohe, M.; Kenney, S. Fatty acid synthase expression is induced by the Epstein-Barr virus immediate-early protein BRLF1 and is required for lytic viral gene expression. J. Virol. 2004, 78, 4197–4206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.W.; Shen, H.; Nobre, L.; Ersing, I.; Paulo, J.A.; Trudeau, S.; Wang, Z.; Smith, N.A.; Ma, Y.; Reinstadler, B.; et al. Epstein-Barr-Virus-Induced One-Carbon Metabolism Drives B Cell Transformation. Cell Metab. 2019, 30, 539–555.e11. [Google Scholar] [CrossRef] [Green Version]
- Delgado, T.; Sanchez, E.L.; Camarda, R.; Lagunoff, M. Global metabolic profiling of infection by an oncogenic virus: KSHV induces and requires lipogenesis for survival of latent infection. PLoS Pathog. 2012, 8, e1002866. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, A.P.; Jacobs, S.R.; Freemerman, A.J.; Makowski, L.; Rathmell, J.C.; Dittmer, D.P.; Damania, B. Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma. Proc. Natl. Acad. Sci. USA 2012, 109, 11818–11823. [Google Scholar] [CrossRef] [Green Version]
- Kuhajda, F.P.; Pizer, E.S.; Li, J.N.; Mani, N.S.; Frehywot, G.L.; Townsend, C.A. Synthesis and antitumor activity of an inhibitor of fatty acid synthase. Proc. Natl. Acad. Sci. USA 2000, 97, 3450–3454. [Google Scholar] [CrossRef]
- Yu, X.; Shahir, A.M.; Sha, J.; Feng, Z.; Eapen, B.; Nithianantham, S.; Das, B.; Karn, J.; Weinberg, A.; Bissada, N.F.; et al. Short-chain fatty acids from periodontal pathogens suppress histone deacetylases, EZH2, and SUV39H1 to promote Kaposi‘s sarcoma-associated herpesvirus replication. J. Virol. 2014, 88, 4466–4479. [Google Scholar] [CrossRef] [Green Version]
- Grillo, M.A.; Colombatto, S. S-adenosylmethionine and its products. Amino Acids 2008, 34, 187–193. [Google Scholar] [CrossRef]
- Lyko, F. The DNA methyltransferase family: A versatile toolkit for epigenetic regulation. Nat. Rev. Genet. 2018, 19, 81–92. [Google Scholar] [CrossRef]
- Rowles, D.L.; Tsai, Y.C.; Greco, T.M.; Lin, A.E.; Li, M.; Yeh, J.; Cristea, I.M. DNA methyltransferase DNMT3A associates with viral proteins and impacts HSV-1 infection. Proteomics 2015, 15, 1968–1982. [Google Scholar] [CrossRef]
- Engdahl, E.; Dunn, N.; Niehusmann, P.; Wideman, S.; Wipfler, P.; Becker, A.J.; Ekstrom, T.J.; Almgren, M.; Fogdell-Hahn, A. Human Herpesvirus 6B Induces Hypomethylation on Chromosome 17p13.3, Correlating with Increased Gene Expression and Virus Integration. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [Green Version]
- Leonard, S.; Wei, W.; Anderton, J.; Vockerodt, M.; Rowe, M.; Murray, P.G.; Woodman, C.B. Epigenetic and transcriptional changes which follow Epstein-Barr virus infection of germinal center B cells and their relevance to the pathogenesis of Hodgkin’s lymphoma. J. Virol. 2011, 85, 9568–9577. [Google Scholar] [CrossRef] [Green Version]
- Paschos, K.; Smith, P.; Anderton, E.; Middeldorp, J.M.; White, R.E.; Allday, M.J. Epstein-barr virus latency in B cells leads to epigenetic repression and CpG methylation of the tumour suppressor gene Bim. PLoS Pathog. 2009, 5, e1000492. [Google Scholar] [CrossRef] [Green Version]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two Epstein-Barr virus (EBV) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor Bim: Clues to the pathogenesis of Burkitt’s lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Hughes, D.J.; Marendy, E.M.; Dickerson, C.A.; Yetming, K.D.; Sample, C.E.; Sample, J.T. Contributions of CTCF and DNA methyltransferases DNMT1 and DNMT3B to Epstein-Barr virus restricted latency. J. Virol. 2012, 86, 1034–1045. [Google Scholar] [CrossRef] [Green Version]
- Tsai, C.L.; Li, H.P.; Lu, Y.J.; Hsueh, C.; Liang, Y.; Chen, C.L.; Tsao, S.W.; Tse, K.P.; Yu, J.S.; Chang, Y.S. Activation of DNA methyltransferase 1 by EBV LMP1 Involves c-Jun NH(2)-terminal kinase signaling. Cancer Res. 2006, 66, 11668–11676. [Google Scholar] [CrossRef] [Green Version]
- Shamay, M.; Krithivas, A.; Zhang, J.; Hayward, S.D. Recruitment of the de novo DNA methyltransferase Dnmt3a by Kaposi’s sarcoma-associated herpesvirus LANA. Proc. Natl. Acad. Sci. USA 2006, 103, 14554–14559. [Google Scholar] [CrossRef] [Green Version]
- Lu, F.; Stedman, W.; Yousef, M.; Renne, R.; Lieberman, P.M. Epigenetic regulation of Kaposi’s sarcoma-associated herpesvirus latency by virus-encoded microRNAs that target Rta and the cellular Rbl2-DNMT pathway. J. Virol. 2010, 84, 2697–2706. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wang, Q.; Feng, Q.; Wang, F.; Yan, Q.; Gao, S.J.; Lu, C. Oncogenic KSHV-encoded interferon regulatory factor upregulates HMGB2 and CMPK1 expression to promote cell invasion by disrupting a complex lncRNA-OIP5-AS1/miR-218-5p network. PLoS Pathog. 2019, 15, e1007578. [Google Scholar] [CrossRef]
- Morera, L.; Lubbert, M.; Jung, M. Targeting histone methyltransferases and demethylases in clinical trials for cancer therapy. Clin. Epigenetics 2016, 8, 57. [Google Scholar] [CrossRef] [Green Version]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Husmann, D.; Gozani, O. Histone lysine methyltransferases in biology and disease. Nat. Struct. Mol. Biol. 2019, 26, 880–889. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Bedford, M.T. Histone arginine methylation. FEBS Lett. 2011, 585, 2024–2031. [Google Scholar] [CrossRef] [Green Version]
- Black, J.C.; Van Rechem, C.; Whetstine, J.R. Histone lysine methylation dynamics: Establishment, regulation, and biological impact. Mol. Cell 2012, 48, 491–507. [Google Scholar] [CrossRef] [Green Version]
- Ichikawa, T.; Okuno, Y.; Sato, Y.; Goshima, F.; Yoshiyama, H.; Kanda, T.; Kimura, H.; Murata, T. Regulation of Epstein-Barr Virus Life Cycle and Cell Proliferation by Histone H3K27 Methyltransferase EZH2 in Akata Cells. mSphere 2018, 3. [Google Scholar] [CrossRef] [Green Version]
- Arbuckle, J.H.; Gardina, P.J.; Gordon, D.N.; Hickman, H.D.; Yewdell, J.W.; Pierson, T.C.; Myers, T.G.; Kristie, T.M. Inhibitors of the Histone Methyltransferases EZH2/1 Induce a Potent Antiviral State and Suppress Infection by Diverse Viral Pathogens. mBio 2017, 8. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Kent, J.R.; Placek, B.; Whelan, K.A.; Hollow, C.M.; Zeng, P.Y.; Fraser, N.W.; Berger, S.L. Trimethylation of histone H3 lysine 4 by Set1 in the lytic infection of human herpes simplex virus 1. J. Virol. 2006, 80, 5740–5746. [Google Scholar] [CrossRef] [Green Version]
- Narayanan, A.; Ruyechan, W.T.; Kristie, T.M. The coactivator host cell factor-1 mediates Set1 and MLL1 H3K4 trimethylation at herpesvirus immediate early promoters for initiation of infection. Proc. Natl. Acad. Sci. USA 2007, 104, 10835–10840. [Google Scholar] [CrossRef] [Green Version]
- Strahan, R.C.; McDowell-Sargent, M.; Uppal, T.; Purushothaman, P.; Verma, S.C. KSHV encoded ORF59 modulates histone arginine methylation of the viral genome to promote viral reactivation. PLoS Pathog. 2017, 13, e1006482. [Google Scholar] [CrossRef]
- Etchegaray, J.P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [Green Version]
- Kaelin, W.G., Jr.; McKnight, S.L. Influence of metabolism on epigenetics and disease. Cell 2013, 153, 56–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [Green Version]
- Ward, P.S.; Patel, J.; Wise, D.R.; Abdel-Wahab, O.; Bennett, B.D.; Coller, H.A.; Cross, J.R.; Fantin, V.R.; Hedvat, C.V.; Perl, A.E.; et al. The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 2010, 17, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Lu, C.; Ward, P.S.; Kapoor, G.S.; Rohle, D.; Turcan, S.; Abdel-Wahab, O.; Edwards, C.R.; Khanin, R.; Figueroa, M.E.; Melnick, A.; et al. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 2012, 483, 474–478. [Google Scholar] [CrossRef] [Green Version]
- Teperino, R.; Schoonjans, K.; Auwerx, J. Histone methyl transferases and demethylases; Can they link metabolism and transcription? Cell Metab. 2010, 12, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Pastor, W.A.; Aravind, L.; Rao, A. TETonic shift: Biological roles of TET proteins in DNA demethylation and transcription. Nat. Rev. Mol. Cell Biol. 2013, 14, 341–356. [Google Scholar] [CrossRef] [Green Version]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef] [Green Version]
- Kohli, R.M.; Zhang, Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature 2013, 502, 472–479. [Google Scholar] [CrossRef] [Green Version]
- Ito, S.; D’Alessio, A.C.; Taranova, O.V.; Hong, K.; Sowers, L.C.; Zhang, Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 2010, 466, 1129–1133. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.; Zhou, M.; Shang, L.; Du, Q.; Li, Y.; Xie, L.; Liu, X.; Tang, M.; Luo, X.; Fan, J.; et al. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics 2019, 9, 2424–2438. [Google Scholar] [CrossRef]
- Dimitrova, E.; Turberfield, A.H.; Klose, R.J. Histone demethylases in chromatin biology and beyond. EMBO Rep. 2015, 16, 1620–1639. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, Y.; Fang, J.; Erdjument-Bromage, H.; Warren, M.E.; Borchers, C.H.; Tempst, P.; Zhang, Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature 2006, 439, 811–816. [Google Scholar] [CrossRef]
- Anand, R.; Marmorstein, R. Structure and mechanism of lysine-specific demethylase enzymes. J. Biol. Chem. 2007, 282, 35425–35429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karytinos, A.; Forneris, F.; Profumo, A.; Ciossani, G.; Battaglioli, E.; Binda, C.; Mattevi, A. A novel mammalian flavin-dependent histone demethylase. J. Biol. Chem. 2009, 284, 17775–17782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Gunther, T.; Buettner, R.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar] [CrossRef]
- Ciccone, D.N.; Su, H.; Hevi, S.; Gay, F.; Lei, H.; Bajko, J.; Xu, G.; Li, E.; Chen, T. KDM1B is a histone H3K4 demethylase required to establish maternal genomic imprints. Nature 2009, 461, 415–418. [Google Scholar] [CrossRef]
- Forneris, F.; Binda, C.; Vanoni, M.A.; Mattevi, A.; Battaglioli, E. Histone demethylation catalysed by LSD1 is a flavin-dependent oxidative process. FEBS Lett. 2005, 579, 2203–2207. [Google Scholar] [CrossRef] [Green Version]
- Janke, R.; Dodson, A.E.; Rine, J. Metabolism and epigenetics. Annu. Rev. Cell Dev. Biol. 2015, 31, 473–496. [Google Scholar] [CrossRef] [Green Version]
- Whetstine, J.R.; Nottke, A.; Lan, F.; Huarte, M.; Smolikov, S.; Chen, Z.; Spooner, E.; Li, E.; Zhang, G.; Colaiacovo, M.; et al. Reversal of histone lysine trimethylation by the JMJD2 family of histone demethylases. Cell 2006, 125, 467–481. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Cho, Y.W.; Yu, L.R.; Yu, H.; Veenstra, T.D.; Ge, K. Identification of JmjC domain-containing UTX and JMJD3 as histone H3 lysine 27 demethylases. Proc. Natl. Acad. Sci. USA 2007, 104, 18439–18444. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Vogel, J.L.; Narayanan, A.; Peng, H.; Kristie, T.M. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 2009, 15, 1312–1317. [Google Scholar] [CrossRef] [PubMed]
- Oh, H.S.; Bryant, K.F.; Nieland, T.J.; Mazumder, A.; Bagul, M.; Bathe, M.; Root, D.E.; Knipe, D.M. A targeted RNA interference screen reveals novel epigenetic factors that regulate herpesviral gene expression. mBio 2014, 5, e01086-01013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocka, J.; Myers, M.P.; Laherty, C.D.; Eisenman, R.N.; Herr, W. Human Sin3 deacetylase and trithorax-related Set1/Ash2 histone H3-K4 methyltransferase are tethered together selectively by the cell-proliferation factor HCF-1. Genes Dev. 2003, 17, 896–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Y.; Vogel, J.L.; Arbuckle, J.H.; Rai, G.; Jadhav, A.; Simeonov, A.; Maloney, D.J.; Kristie, T.M. Targeting the JMJD2 histone demethylases to epigenetically control herpesvirus infection and reactivation from latency. Sci. Transl. Med. 2013, 5, 167ra165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.M.; Quenelle, D.C.; Cardin, R.D.; Vogel, J.L.; Clement, C.; Bravo, F.J.; Foster, T.P.; Bosch-Marce, M.; Raja, P.; Lee, J.S.; et al. Inhibition of LSD1 reduces herpesvirus infection, shedding, and recurrence by promoting epigenetic suppression of viral genomes. Sci. Transl. Med. 2014, 6, 265ra169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.R.; Wong, L.Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010, 6, e1001013. [Google Scholar] [CrossRef] [Green Version]
- Verdin, E.; Ott, M. 50 years of protein acetylation: From gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol. 2015, 16, 258–264. [Google Scholar] [CrossRef]
- Lee, K.K.; Workman, J.L. Histone acetyltransferase complexes: One size doesn’t fit all. Nat. Rev. Mol. Cell Biol. 2007, 8, 284–295. [Google Scholar] [CrossRef]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [Green Version]
- Chang, L.K.; Liu, S.T. Activation of the BRLF1 promoter and lytic cycle of Epstein-Barr virus by histone acetylation. Nucleic Acids Res 2000, 28, 3918–3925. [Google Scholar] [CrossRef]
- Jenkins, P.J.; Binne, U.K.; Farrell, P.J. Histone acetylation and reactivation of Epstein-Barr virus from latency. J. Virol. 2000, 74, 710–720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Grossman, S.R.; Kieff, E. Epstein-Barr virus nuclear protein 2 interacts with p300, CBP, and PCAF histone acetyltransferases in activation of the LMP1 promoter. Proc. Natl. Acad. Sci. USA 2000, 97, 430–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kornberg, R.D.; Lorch, Y. Twenty-five years of the nucleosome, fundamental particle of the eukaryote chromosome. Cell 1999, 98, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Alazard, N.; Gruffat, H.; Hiriart, E.; Sergeant, A.; Manet, E. Differential hyperacetylation of histones H3 and H4 upon promoter-specific recruitment of EBNA2 in Epstein-Barr virus chromatin. J. Virol. 2003, 77, 8166–8172. [Google Scholar] [CrossRef] [Green Version]
- Choi, K.C.; Jung, M.G.; Lee, Y.H.; Yoon, J.C.; Kwon, S.H.; Kang, H.B.; Kim, M.J.; Cha, J.H.; Kim, Y.J.; Jun, W.J.; et al. Epigallocatechin-3-gallate, a histone acetyltransferase inhibitor, inhibits EBV-induced B lymphocyte transformation via suppression of RelA acetylation. Cancer Res. 2009, 69, 583–592. [Google Scholar] [CrossRef] [Green Version]
- Wakil, S.J.; Abu-Elheiga, L.A. Fatty acid metabolism: Target for metabolic syndrome. J. Lipid Res. 2009, 50, S138–S143. [Google Scholar] [CrossRef] [Green Version]
- Spencer, C.M.; Schafer, X.L.; Moorman, N.J.; Munger, J. Human cytomegalovirus induces the activity and expression of acetyl-coenzyme A carboxylase, a fatty acid biosynthetic enzyme whose inhibition attenuates viral replication. J. Virol. 2011, 85, 5814–5824. [Google Scholar] [CrossRef] [Green Version]
- Galdieri, L.; Vancura, A. Acetyl-CoA carboxylase regulates global histone acetylation. J. Biol. Chem. 2012, 287, 23865–23876. [Google Scholar] [CrossRef] [Green Version]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef]
- Imai, S.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000, 403, 795–800. [Google Scholar] [CrossRef]
- Landry, J.; Sutton, A.; Tafrov, S.T.; Heller, R.C.; Stebbins, J.; Pillus, L.; Sternglanz, R. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proc. Natl. Acad. Sci. USA 2000, 97, 5807–5811. [Google Scholar] [CrossRef] [Green Version]
- Feldman, J.L.; Dittenhafer-Reed, K.E.; Denu, J.M. Sirtuin catalysis and regulation. J. Biol. Chem. 2012, 287, 42419–42427. [Google Scholar] [CrossRef] [Green Version]
- Koyuncu, E.; Budayeva, H.G.; Miteva, Y.V.; Ricci, D.P.; Silhavy, T.J.; Shenk, T.; Cristea, I.M. Sirtuins are evolutionarily conserved viral restriction factors. mBio 2014, 5. [Google Scholar] [CrossRef] [Green Version]
- Solomon, J.M.; Pasupuleti, R.; Xu, L.; McDonagh, T.; Curtis, R.; DiStefano, P.S.; Huber, L.J. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol. Cell. Biol. 2006, 26, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.; He, M.; Zhou, F.; Ye, F.; Gao, S.J. Activation of Kaposi’s sarcoma-associated herpesvirus (KSHV) by inhibitors of class III histone deacetylases: Identification of sirtuin 1 as a regulator of the KSHV life cycle. J. Virol. 2014, 88, 6355–6367. [Google Scholar] [CrossRef] [Green Version]
- Lomonte, P.; Thomas, J.; Texier, P.; Caron, C.; Khochbin, S.; Epstein, A.L. Functional interaction between class II histone deacetylases and ICP0 of herpes simplex virus type 1. J. Virol. 2004, 78, 6744–6757. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Roizman, B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. USA 2007, 104, 17134–17139. [Google Scholar] [CrossRef] [Green Version]
- Gu, H.; Liang, Y.; Mandel, G.; Roizman, B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 7571–7576. [Google Scholar] [CrossRef] [Green Version]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar] [CrossRef] [Green Version]
- Cliffe, A.R.; Knipe, D.M. Herpes simplex virus ICP0 promotes both histone removal and acetylation on viral DNA during lytic infection. J. Virol. 2008, 82, 12030–12038. [Google Scholar] [CrossRef] [Green Version]
- Coleman, H.M.; Connor, V.; Cheng, Z.S.C.; Grey, F.; Preston, C.M.; Efstathiou, S. Histone modifications associated with herpes simplex virus type 1 genomes during quiescence and following ICP0-mediated de-repression. J. Gen. Virol. 2008, 89, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Danaher, R.J.; Jacob, R.J.; Steiner, M.R.; Allen, W.R.; Hill, J.M.; Miller, C.S. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J. Neurovirol. 2005, 11, 306–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perrine, S.P.; Hermine, O.; Small, T.; Suarez, F.; O’Reilly, R.; Boulad, F.; Fingeroth, J.; Askin, M.; Levy, A.; Mentzer, S.J.; et al. A phase 1/2 trial of arginine butyrate and ganciclovir in patients with Epstein-Barr virus-associated lymphoid malignancies. Blood 2007, 109, 2571–2578. [Google Scholar] [CrossRef] [Green Version]
- Ghosh, S.K.; Perrine, S.P.; Williams, R.M.; Faller, D.V. Histone deacetylase inhibitors are potent inducers of gene expression in latent EBV and sensitize lymphoma cells to nucleoside antiviral agents. Blood 2012, 119, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.C.; Fischle, W.; Verdin, E.; Sinclair, J.H. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 2002, 21, 1112–1120. [Google Scholar] [CrossRef] [Green Version]
- Eagle, H. Nutrition needs of mammalian cells in tissue culture. Science 1955, 122, 501–514. [Google Scholar] [CrossRef]
- Zhang, J.; Pavlova, N.N.; Thompson, C.B. Cancer cell metabolism: The essential role of the nonessential amino acid, glutamine. EMBO J 2017, 36, 1302–1315. [Google Scholar] [CrossRef] [Green Version]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.F.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015, 35, S129–S150. [Google Scholar] [CrossRef] [Green Version]
- Chambers, J.W.; Maguire, T.G.; Alwine, J.C. Glutamine metabolism is essential for human cytomegalovirus infection. J. Virol. 2010, 84, 1867–1873. [Google Scholar] [CrossRef] [Green Version]
- Sanchez, E.L.; Carroll, P.A.; Thalhofer, A.B.; Lagunoff, M. Latent KSHV Infected Endothelial Cells Are Glutamine Addicted and Require Glutaminolysis for Survival. PLoS Pathog. 2015, 11, e1005052. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pei, Y.; Robertson, E.S. The Crosstalk of Epigenetics and Metabolism in Herpesvirus Infection. Viruses 2020, 12, 1377. https://doi.org/10.3390/v12121377
Pei Y, Robertson ES. The Crosstalk of Epigenetics and Metabolism in Herpesvirus Infection. Viruses. 2020; 12(12):1377. https://doi.org/10.3390/v12121377
Chicago/Turabian StylePei, Yonggang, and Erle S. Robertson. 2020. "The Crosstalk of Epigenetics and Metabolism in Herpesvirus Infection" Viruses 12, no. 12: 1377. https://doi.org/10.3390/v12121377
APA StylePei, Y., & Robertson, E. S. (2020). The Crosstalk of Epigenetics and Metabolism in Herpesvirus Infection. Viruses, 12(12), 1377. https://doi.org/10.3390/v12121377