Comparative Analysis of the Circular and Highly Asymmetrical Marseilleviridae Genomes


Abstract
1. Introduction
2. Materials and Methods
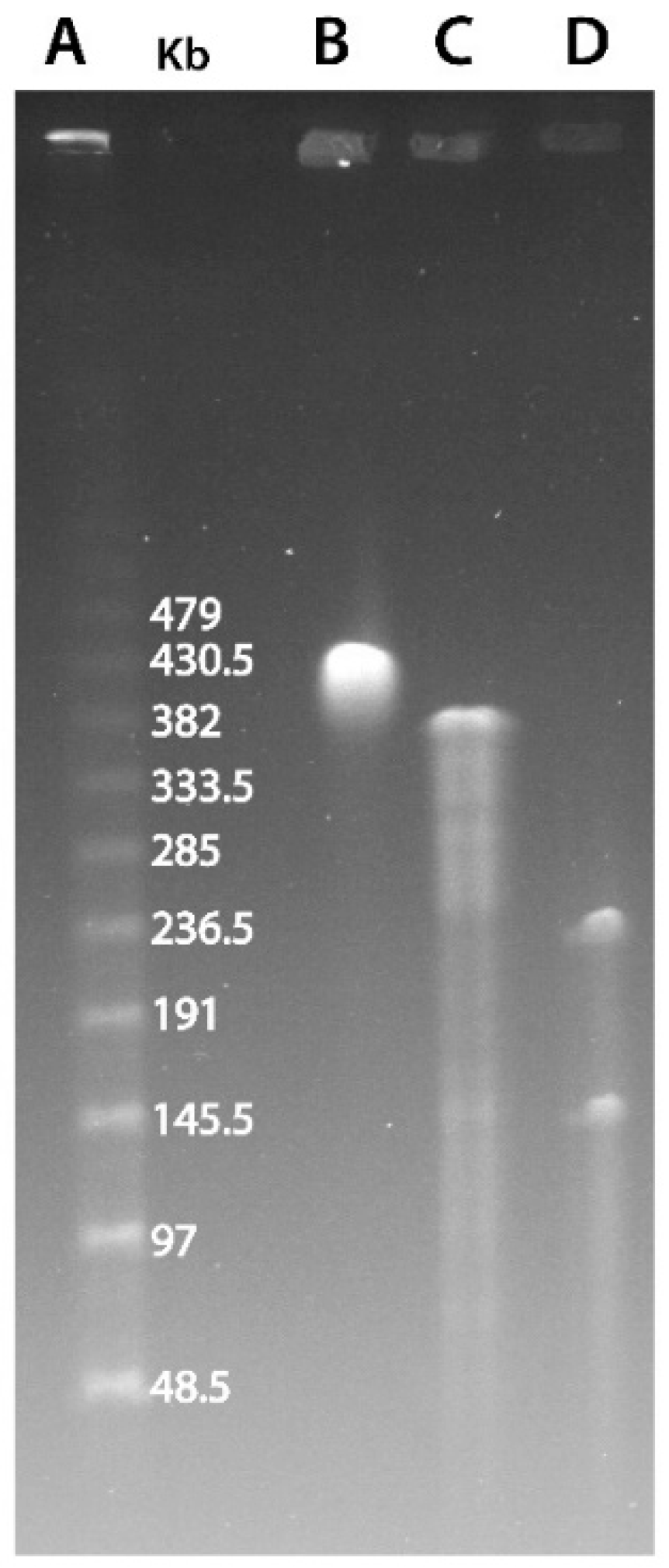
2.1. Pulse-Field Gel Electrophoresis
2.2. Genome Analysis
2.3. Homologous Proteins Clusering and Pangenome Analysis
2.4. Nucleotide Biais Composition
2.5. Transcriptomic Data Analyses
2.6. Phylogeny and Selection Pressure Analysis
3. Results
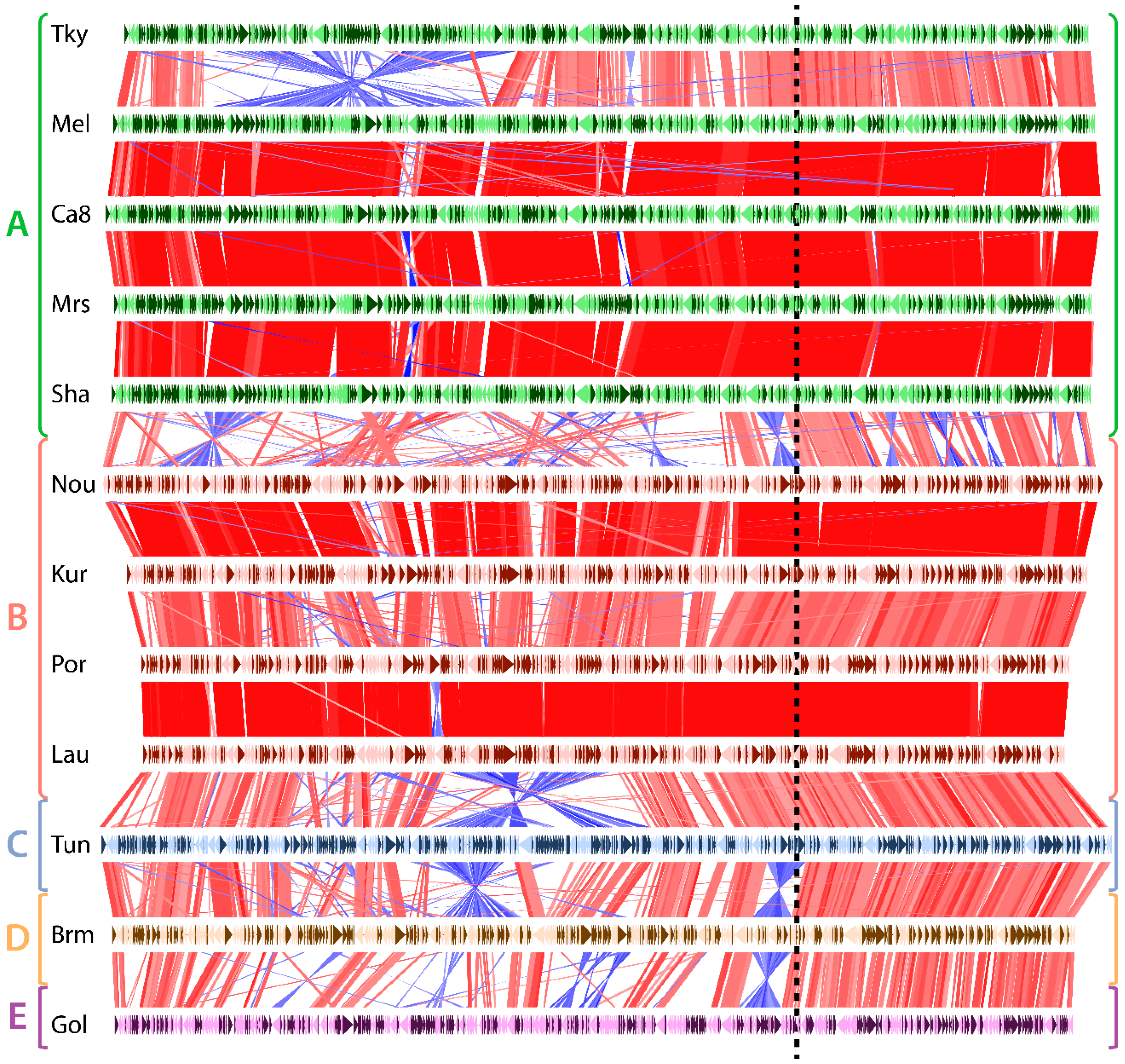
3.1. Marseilleviridae Genomes Are Circular
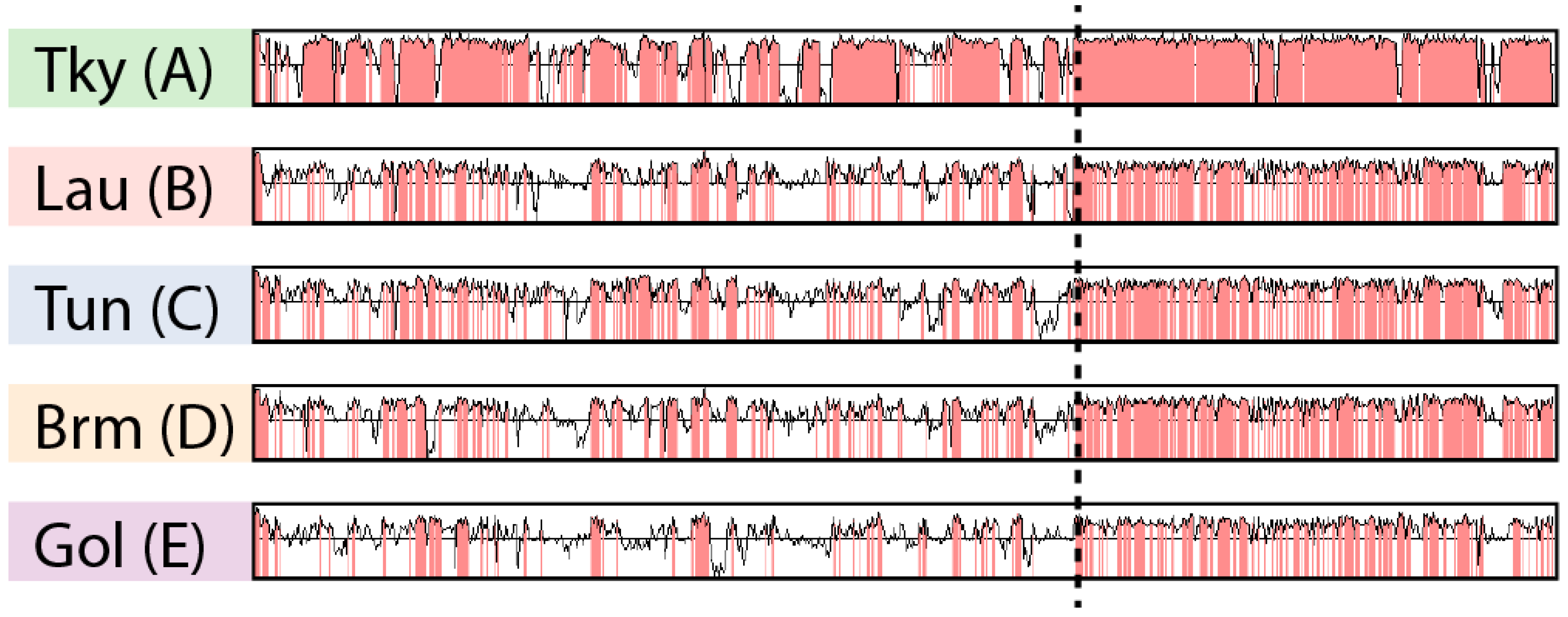
3.2. Asymetry in Sequence Conservation along the Genomes
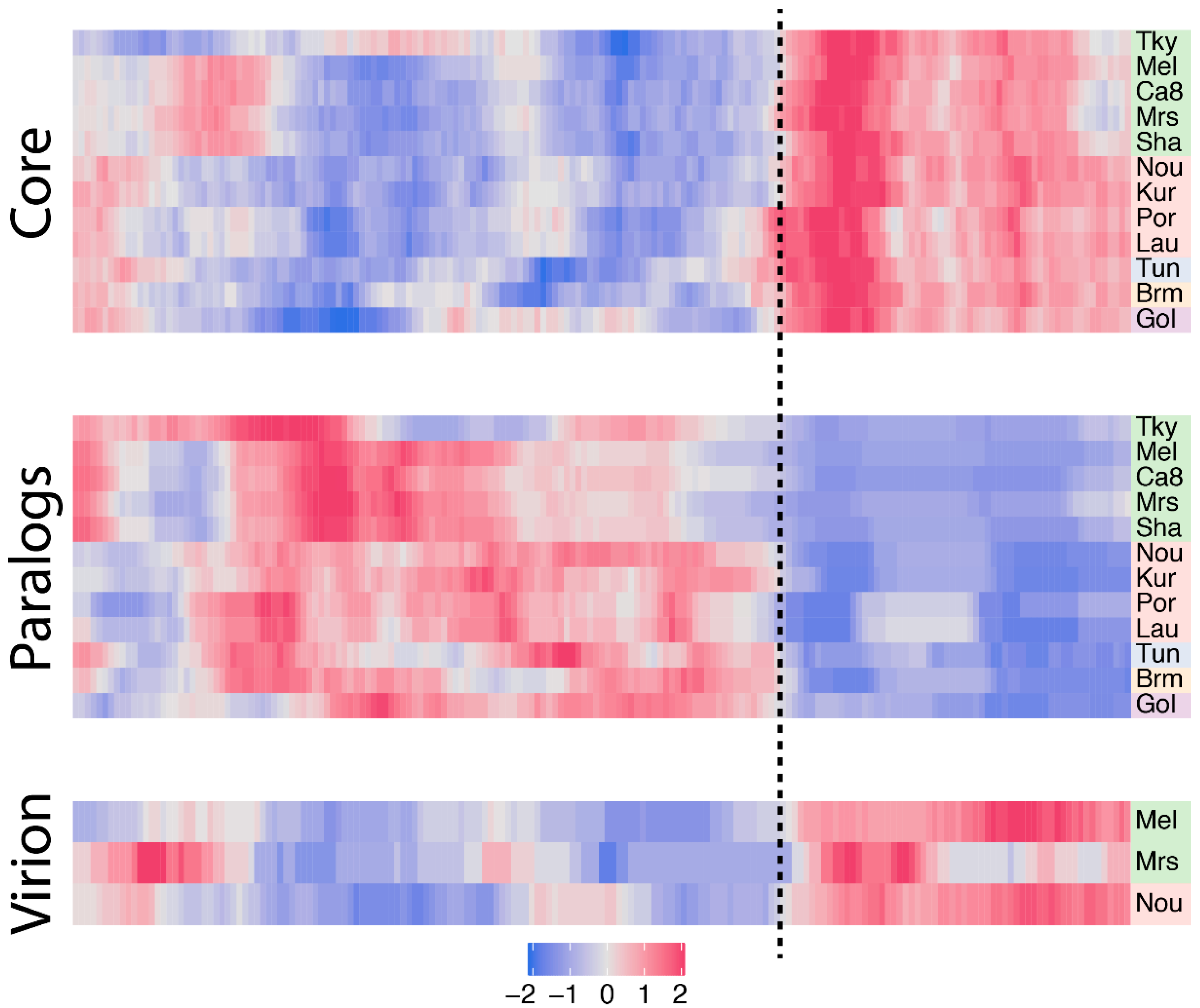
3.3. Biaised Distribution of Core Genes
3.4. Biaised Distribution of Virion-Associated Proteins and Late-Expressed Transcripts
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Boyer, M.; Yutin, N.; Pagnier, I.; Barrassi, L.; Fournous, G.; Espinosa, L.; Robert, C.; Azza, S.; Sun, S.; Rossmann, M.G.; et al. Giant Marseillevirus highlights the role of amoebae as a melting pot in emergence of chimeric microorganisms. Proc. Natl. Acad. Sci. USA 2009, 106, 21848–21853. [Google Scholar] [CrossRef]
- Thomas, V.; Bertelli, C.; Collyn, F.; Casson, N.; Telenti, A.; Goesmann, A.; Croxatto, A.; Greub, G. Lausannevirus, a giant amoebal virus encoding histone doublets. Environ. Microbiol. 2011, 13, 1454–1466. [Google Scholar] [CrossRef]
- Aherfi, S.; Pagnier, I.; Fournous, G.; Raoult, D.; La Scola, B.; Colson, P. Complete genome sequence of Cannes 8 virus, a new member of the proposed family “Marseilleviridae”. Virus Genes 2013, 47, 550–555. [Google Scholar] [CrossRef] [PubMed]
- Boughalmi, M.; Pagnier, I.; Aherfi, S.; Colson, P.; Raoult, D.; La Scola, B. First isolation of a Marseillevirus in the Diptera Syrphidae Eristalis tenax. Intervirology 2013, 56, 386–394. [Google Scholar] [CrossRef]
- Aherfi, S.; Boughalmi, M.; Pagnier, I.; Fournous, G.; La Scola, B.; Raoult, D.; Colson, P. Complete genome sequence of Tunisvirus, a new member of the proposed family Marseilleviridae. Arch. Virol. 2014, 159, 2349–2358. [Google Scholar] [CrossRef]
- Dornas, F.P.; Assis, F.L.; Aherfi, S.; Arantes, T.; Abrahão, J.S.; Colson, P.; La Scola, B. A Brazilian Marseillevirus Is the Founding Member of a Lineage in Family Marseilleviridae. Viruses 2016, 8, 76. [Google Scholar] [CrossRef]
- Doutre, G.; Philippe, N.; Abergel, C.; Claverie, J.-M. Genome analysis of the first Marseilleviridae representative from Australia indicates that most of its genes contribute to virus fitness. J. Virol. 2014, 88, 14340–14349. [Google Scholar] [CrossRef]
- Doutre, G.; Arfib, B.; Rochette, P.; Claverie, J.-M.; Bonin, P.; Abergel, C. Complete Genome Sequence of a New Member of the Marseilleviridae Recovered from the Brackish Submarine Spring in the Cassis Port-Miou Calanque, France. Genome Announc. 2015, 3. [Google Scholar] [CrossRef] [PubMed]
- Takemura, M. Draft Genome Sequence of Tokyovirus, a Member of the Family Marseilleviridae Isolated from the Arakawa River of Tokyo, Japan. Genome Announc. 2016, 4. [Google Scholar] [CrossRef]
- Fabre, E.; Jeudy, S.; Santini, S.; Legendre, M.; Trauchessec, M.; Couté, Y.; Claverie, J.-M.; Abergel, C. Noumeavirus replication relies on a transient remote control of the host nucleus. Nat. Commun. 2017, 8, 15087. [Google Scholar] [CrossRef]
- Dos Santos, R.N.; Campos, F.S.; Medeiros de Albuquerque, N.R.; Finoketti, F.; Côrrea, R.A.; Cano-Ortiz, L.; Assis, F.L.; Arantes, T.S.; Roehe, P.M.; Franco, A.C. A new marseillevirus isolated in Southern Brazil from Limnoperna fortunei. Sci. Rep. 2016, 6, 35237. [Google Scholar] [CrossRef]
- Chatterjee, A.; Kondabagil, K. Complete genome sequence of Kurlavirus, a novel member of the family Marseilleviridae isolated in Mumbai, India. Arch. Virol. 2017, 162, 3243–3245. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 16 September 2020).
- International Committee on Taxonomy of Viruses Executive Committee. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef]
- Aoki, K.; Hagiwara, R.; Akashi, M.; Sasaki, K.; Murata, K.; Ogata, H.; Takemura, M. Fifteen Marseilleviruses Newly Isolated From Three Water Samples in Japan Reveal Local Diversity of Marseilleviridae. Front. Microbiol. 2019, 10, 1152. [Google Scholar] [CrossRef]
- Bäckström, D.; Yutin, N.; Jørgensen, S.L.; Dharamshi, J.; Homa, F.; Zaremba-Niedwiedzka, K.; Spang, A.; Wolf, Y.I.; Koonin, E.V.; Ettema, T.J.G. Virus Genomes from Deep Sea Sediments Expand the Ocean Megavirome and Support Independent Origins of Viral Gigantism. mBio 2019, 10, e02497-18. [Google Scholar] [CrossRef]
- Jeudy, S.; Rigou, S.; Alempic, J.-M.; Claverie, J.-M.; Abergel, C.; Legendre, M. The DNA methylation landscape of giant viruses. Nat. Commun. 2020, 11, 2657. [Google Scholar] [CrossRef]
- Rodrigues, R.A.L.; Louazani, A.C.; Picorelli, A.; Oliveira, G.P.; Lobo, F.P.; Colson, P.; La Scola, B.; Abrahão, J.S. Analysis of a Marseillevirus Transcriptome Reveals Temporal Gene Expression Profile and Host Transcriptional Shift. Front. Microbiol. 2020, 11, 651. [Google Scholar] [CrossRef]
- Nurk, S.; Meleshko, D.; Korobeynikov, A.; Pevzner, P.A. metaSPAdes: A new versatile metagenomic assembler. Genome Res. 2017, 27, 824–834. [Google Scholar] [CrossRef]
- Besemer, J.; Lomsadze, A.; Borodovsky, M. GeneMarkS: A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001, 29, 2607–2618. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Carver, T.J.; Rutherford, K.M.; Berriman, M.; Rajandream, M.-A.; Barrell, B.G.; Parkhill, J. ACT: The Artemis Comparison Tool. Bioinformatics 2005, 21, 3422–3423. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef]
- Sievers, F.; Higgins, D.G. Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 2014, 1079, 105–116. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Zhao, Y.; Jia, X.; Yang, J.; Ling, Y.; Zhang, Z.; Yu, J.; Wu, J.; Xiao, J. PanGP: A tool for quickly analyzing bacterial pan-genome profile. Bioinformatics 2014, 30, 1297–1299. [Google Scholar] [CrossRef]
- Snipen, L.; Liland, K.H. Micropan: An R-package for microbial pan-genomics. BMC Bioinform. 2015, 16, 79. [Google Scholar] [CrossRef]
- Necşulea, A.; Lobry, J.R. A new method for assessing the effect of replication on DNA base composition asymmetry. Mol. Biol. Evol. 2007, 24, 2169–2179. [Google Scholar] [CrossRef]
- Vera Alvarez, R.; Pongor, L.S.; Mariño-Ramírez, L.; Landsman, D. TPMCalculator: One-step software to quantify mRNA abundance of genomic features. Bioinformatics 2019, 35, 1960–1962. [Google Scholar] [CrossRef]
- Gu, Z.; Eils, R.; Schlesner, M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016, 32, 2847–2849. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Serra, F.; Bork, P. ETE 3: Reconstruction, Analysis, and Visualization of Phylogenomic Data. Mol. Biol. Evol. 2016, 33, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Barton, B.M.; Harding, G.P.; Zuccarelli, A.J. A general method for detecting and sizing large plasmids. Anal. Biochem. 1995, 226, 235–240. [Google Scholar] [CrossRef]
- Lee, I.; Kim, Y.O.; Park, S.-C.; Chun, J. OrthoANI: An Improved Algorithm and Software for Calculating Average Nucleotide Identity. Available online: http://pubmed.ncbi.nlm.nih.gov/26585518/ (accessed on 21 September 2020).
- Brudno, M.; Malde, S.; Poliakov, A.; Do, C.B.; Couronne, O.; Dubchak, I.; Batzoglou, S. Glocal alignment: Finding rearrangements during alignment. Bioinformatics 2003, 19, i54–i62. [Google Scholar] [CrossRef] [PubMed]
- Nikolaou, C.; Almirantis, Y. A study on the correlation of nucleotide skews and the positioning of the origin of replication: Different modes of replication in bacterial species. Nucleic Acids Res. 2005, 33, 6816–6822. [Google Scholar] [CrossRef]
- Tettelin, H.; Riley, D.; Cattuto, C.; Medini, D. Comparative genomics: The bacterial pan-genome. Curr. Opin. Microbiol. 2008, 11, 472–477. [Google Scholar] [CrossRef]
- Geballa-Koukoulas, K.; Boudjemaa, H.; Andreani, J.; La Scola, B.; Blanc, G. Comparative Genomics Unveils Regionalized Evolution of the Faustovirus Genomes. Viruses 2020, 12, 577. [Google Scholar] [CrossRef]
- Legendre, M.; Bartoli, J.; Shmakova, L.; Jeudy, S.; Labadie, K.; Adrait, A.; Lescot, M.; Poirot, O.; Bertaux, L.; Bruley, C.; et al. Thirty-thousand-year-old distant relative of giant icosahedral DNA viruses with a pandoravirus morphology. Proc. Natl. Acad. Sci. USA 2014, 111, 4274–4279. [Google Scholar] [CrossRef]
- Andreani, J.; Aherfi, S.; Bou Khalil, J.Y.; Di Pinto, F.; Bitam, I.; Raoult, D.; Colson, P.; La Scola, B. Cedratvirus, a Double-Cork Structured Giant Virus, is a Distant Relative of Pithoviruses. Viruses 2016, 8, 300. [Google Scholar] [CrossRef]
- Andreani, J.; Khalil, J.Y.B.; Baptiste, E.; Hasni, I.; Michelle, C.; Raoult, D.; Levasseur, A.; La Scola, B. Orpheovirus IHUMI-LCC2: A New Virus among the Giant Viruses. Front. Microbiol. 2017, 8, 2643. [Google Scholar] [CrossRef]
- Raoult, D.; Audic, S.; Robert, C.; Abergel, C.; Renesto, P.; Ogata, H.; La Scola, B.; Suzan, M.; Claverie, J.-M. The 1.2-megabase genome sequence of Mimivirus. Science 2004, 306, 1344–1350. [Google Scholar] [CrossRef]
- Philippe, N.; Legendre, M.; Doutre, G.; Couté, Y.; Poirot, O.; Lescot, M.; Arslan, D.; Seltzer, V.; Bertaux, L.; Bruley, C.; et al. Pandoraviruses: Amoeba viruses with genomes up to 2.5 Mb reaching that of parasitic eukaryotes. Science 2013, 341, 281–286. [Google Scholar] [CrossRef]
- Legendre, M.; Lartigue, A.; Bertaux, L.; Jeudy, S.; Bartoli, J.; Lescot, M.; Alempic, J.-M.; Ramus, C.; Bruley, C.; Labadie, K.; et al. In-depth study of Mollivirus sibericum, a new 30,000-y-old giant virus infecting Acanthamoeba. Proc. Natl. Acad. Sci. USA 2015, 112, E5327–E5335. [Google Scholar] [CrossRef]
- Andreani, J.; Khalil, J.Y.B.; Sevvana, M.; Benamar, S.; Di Pinto, F.; Bitam, I.; Colson, P.; Klose, T.; Rossmann, M.G.; Raoult, D.; et al. Pacmanvirus, a New Giant Icosahedral Virus at the Crossroads between Asfarviridae and Faustoviruses. J. Virol. 2017, 91. [Google Scholar] [CrossRef]
- Yoshikawa, G.; Blanc-Mathieu, R.; Song, C.; Kayama, Y.; Mochizuki, T.; Murata, K.; Ogata, H.; Takemura, M. Medusavirus, a Novel Large DNA Virus Discovered from Hot Spring Water. J. Virol. 2019, 93, e02130-18. [Google Scholar] [CrossRef]
- Dillingham, M.S.; Kowalczykowski, S.C. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 2008, 72, 642–671. [Google Scholar] [CrossRef]
- Murphy, K.C. Lambda Gam protein inhibits the helicase and chi-stimulated recombination activities of Escherichia coli RecBCD enzyme. J. Bacteriol. 1991, 173, 5808–5821. [Google Scholar] [CrossRef]
- Schmitz-Esser, S.; Toenshoff, E.R.; Haider, S.; Heinz, E.; Hoenninger, V.M.; Wagner, M.; Horn, M. Diversity of bacterial endosymbionts of environmental acanthamoeba isolates. Appl. Environ. Microbiol. 2008, 74, 5822–5831. [Google Scholar] [CrossRef]
- Legendre, M.; Fabre, E.; Poirot, O.; Jeudy, S.; Lartigue, A.; Alempic, J.-M.; Beucher, L.; Philippe, N.; Bertaux, L.; Christo-Foroux, E.; et al. Diversity and evolution of the emerging Pandoraviridae family. Nat. Commun. 2018, 9, 2285. [Google Scholar] [CrossRef]
- Christo-Foroux, E.; Alempic, J.-M.; Lartigue, A.; Santini, S.; Labadie, K.; Legendre, M.; Abergel, C.; Claverie, J.-M. Characterization of Mollivirus kamchatka, the First Modern Representative of the Proposed Molliviridae Family of Giant Viruses. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Arslan, D.; Legendre, M.; Seltzer, V.; Abergel, C.; Claverie, J.-M. Distant Mimivirus relative with a larger genome highlights the fundamental features of Megaviridae. Proc. Natl. Acad. Sci. USA 2011, 108, 17486–17491. [Google Scholar] [CrossRef]
- McLysaght, A.; Baldi, P.F.; Gaut, B.S. Extensive gene gain associated with adaptive evolution of poxviruses. Proc. Natl. Acad. Sci. USA 2003, 100, 15655–15660. [Google Scholar] [CrossRef]
- Suhre, K.; Audic, S.; Claverie, J.-M. Mimivirus gene promoters exhibit an unprecedented conservation among all eukaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 14689–14693. [Google Scholar] [CrossRef]
- Legendre, M.; Audic, S.; Poirot, O.; Hingamp, P.; Seltzer, V.; Byrne, D.; Lartigue, A.; Lescot, M.; Bernadac, A.; Poulain, J.; et al. mRNA deep sequencing reveals 75 new genes and a complex transcriptional landscape in Mimivirus. Genome Res. 2010, 20, 664–674. [Google Scholar] [CrossRef]
- Lato, D.F.; Golding, G.B. Spatial Patterns of Gene Expression in Bacterial Genomes. J. Mol. Evol. 2020, 88, 510–520. [Google Scholar] [CrossRef]
- Couturier, E.; Rocha, E.P.C. Replication-associated gene dosage effects shape the genomes of fast-growing bacteria but only for transcription and translation genes. Mol. Microbiol. 2006, 59, 1506–1518. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.P.C. The replication-related organization of bacterial genomes. Microbiology 2004, 150, 1609–1627. [Google Scholar] [CrossRef]
- Kelman, L.M.; Kelman, Z. Multiple origins of replication in archaea. Trends Microbiol. 2004, 12, 399–401. [Google Scholar] [CrossRef]
- Senkevich, T.G.; Bruno, D.; Martens, C.; Porcella, S.F.; Wolf, Y.I.; Moss, B. Mapping vaccinia virus DNA replication origins at nucleotide level by deep sequencing. Proc. Natl. Acad. Sci. USA 2015, 112, 10908–10913. [Google Scholar] [CrossRef]
- Schulz, F.; Roux, S.; Paez-Espino, D.; Jungbluth, S.; Walsh, D.A.; Denef, V.J.; McMahon, K.D.; Konstantinidis, K.T.; Eloe-Fadrosh, E.A.; Kyrpides, N.C.; et al. Giant virus diversity and host interactions through global metagenomics. Nature 2020, 578, 432–436. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Core | Strain-Specific | Single Copy | Paralogs | |
|---|---|---|---|---|
| Tokyovirus | 269 (55%) | 35 (7%) | 396 (81%) | 95 (19%) |
| Melbournevirus | 265 (52%) | 6 (1%) | 409 (81%) | 96 (19%) |
| Cannes 8 virus | 268 (53%) | 3 (1%) | 407 (80%) | 103 (20%) |
| Marseillevirus | 264 (52%) | 7 (1%) | 402 (79%) | 107 (21%) |
| Marseillevirus shanghai | 266 (53%) | 3 (1%) | 404 (80%) | 101 (20%) |
| Noumeavirus | 277 (55%) | 16 (3%) | 394 (78%) | 113 (22%) |
| Kurlavirus | 272 (55%) | 12 (2%) | 381 (77%) | 114 (23%) |
| Port-miou virus | 269 (57%) | 8 (2%) | 383 (82%) | 95 (18%) |
| Lausannevirus | 266 (58%) | 3 (1%) | 375 (81%) | 86 (19%) |
| Tunisvirus | 281 (52%) | 31 (6%) | 385 (71%) | 155 (29%) |
| Brazilian marseillevirus | 272 (56%) | 13 (3%) | 373 (77%) | 114 (23%) |
| Golden marseillevirus | 282 (52%) | 76 (14%) | 373 (69%) | 170 (31%) |
| Early Expressed Genes Counts (%) | Intermediate Expressed Genes Counts (%) | Late Expressed Genes Counts (%) | * Maximal Expression (Mean ± SD) | * Total Expression (Mean ± SD) | |
|---|---|---|---|---|---|
| Core-region | 11 (7%) | 39 (23%) | 116 (70%) | 7.4 ± 1.3 | 52 ± 9.8 |
| Other region | 50 (15%) | 143 (42%) | 150 (44%) | 7.3 ± 1.3 | 51 ± 9.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanca, L.; Christo-Foroux, E.; Rigou, S.; Legendre, M. Comparative Analysis of the Circular and Highly Asymmetrical Marseilleviridae Genomes. Viruses 2020, 12, 1270. https://doi.org/10.3390/v12111270
Blanca L, Christo-Foroux E, Rigou S, Legendre M. Comparative Analysis of the Circular and Highly Asymmetrical Marseilleviridae Genomes. Viruses. 2020; 12(11):1270. https://doi.org/10.3390/v12111270
Chicago/Turabian StyleBlanca, Léo, Eugène Christo-Foroux, Sofia Rigou, and Matthieu Legendre. 2020. "Comparative Analysis of the Circular and Highly Asymmetrical Marseilleviridae Genomes" Viruses 12, no. 11: 1270. https://doi.org/10.3390/v12111270
APA StyleBlanca, L., Christo-Foroux, E., Rigou, S., & Legendre, M. (2020). Comparative Analysis of the Circular and Highly Asymmetrical Marseilleviridae Genomes. Viruses, 12(11), 1270. https://doi.org/10.3390/v12111270