Portable Rabies Virus Sequencing in Canine Rabies Endemic Countries Using the Oxford Nanopore MinION

 ,
,  , , , , , , , and
, , , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples
2.2. Nanopore Sequencing
2.3. Sequence Data Analysis
2.4. Manual Homopolymer Indel Correction
2.5. Analysis of Sequence Accuracy
2.6. Sanger Sequencing
2.7. Consensus Accuracy
2.8. Portable Sequencing
2.9. Phylogenetic Analysis
2.10. Data Visualization
2.11. Cost Estimate
3. Results
3.1. Study Design
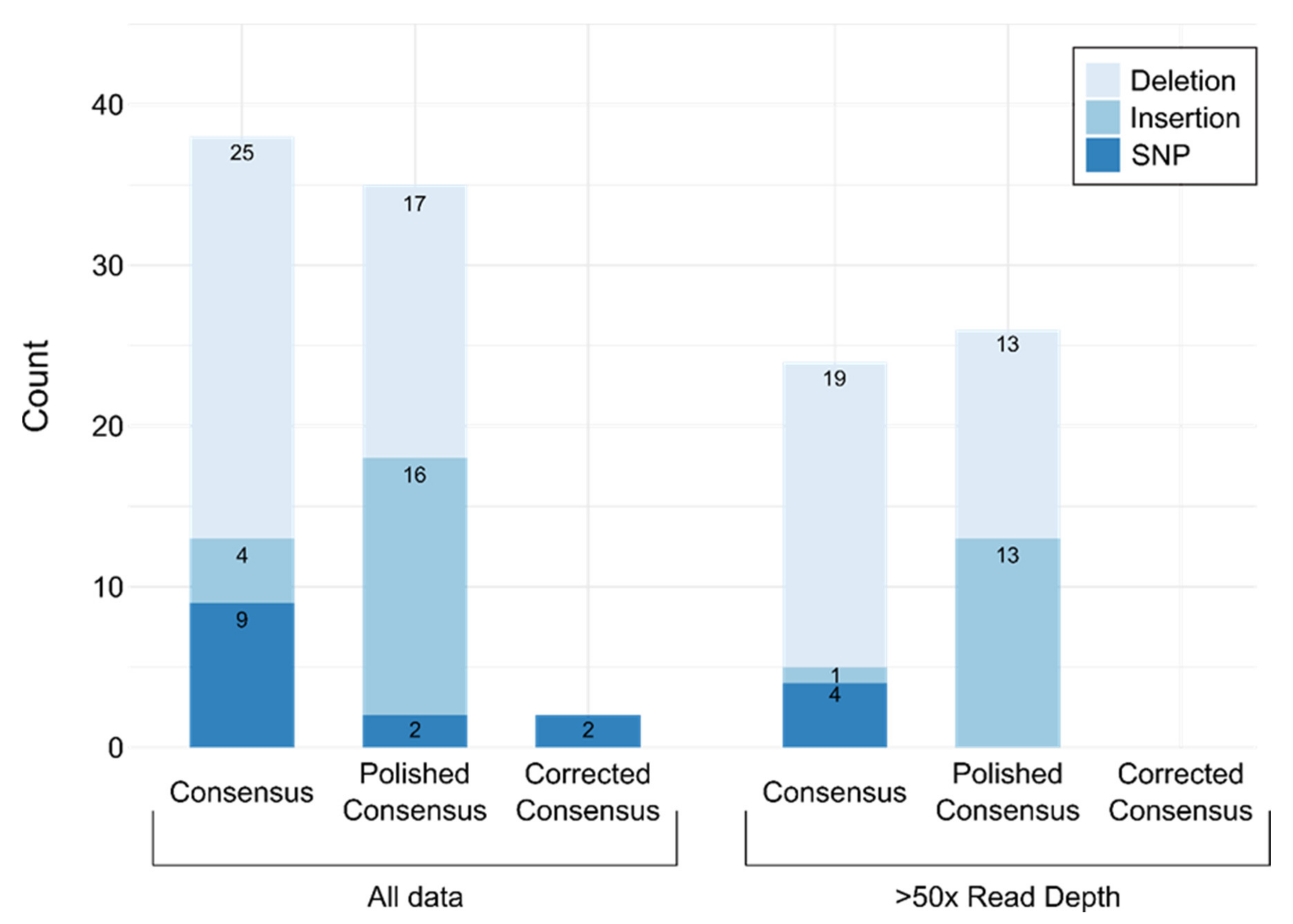
3.2. Validation
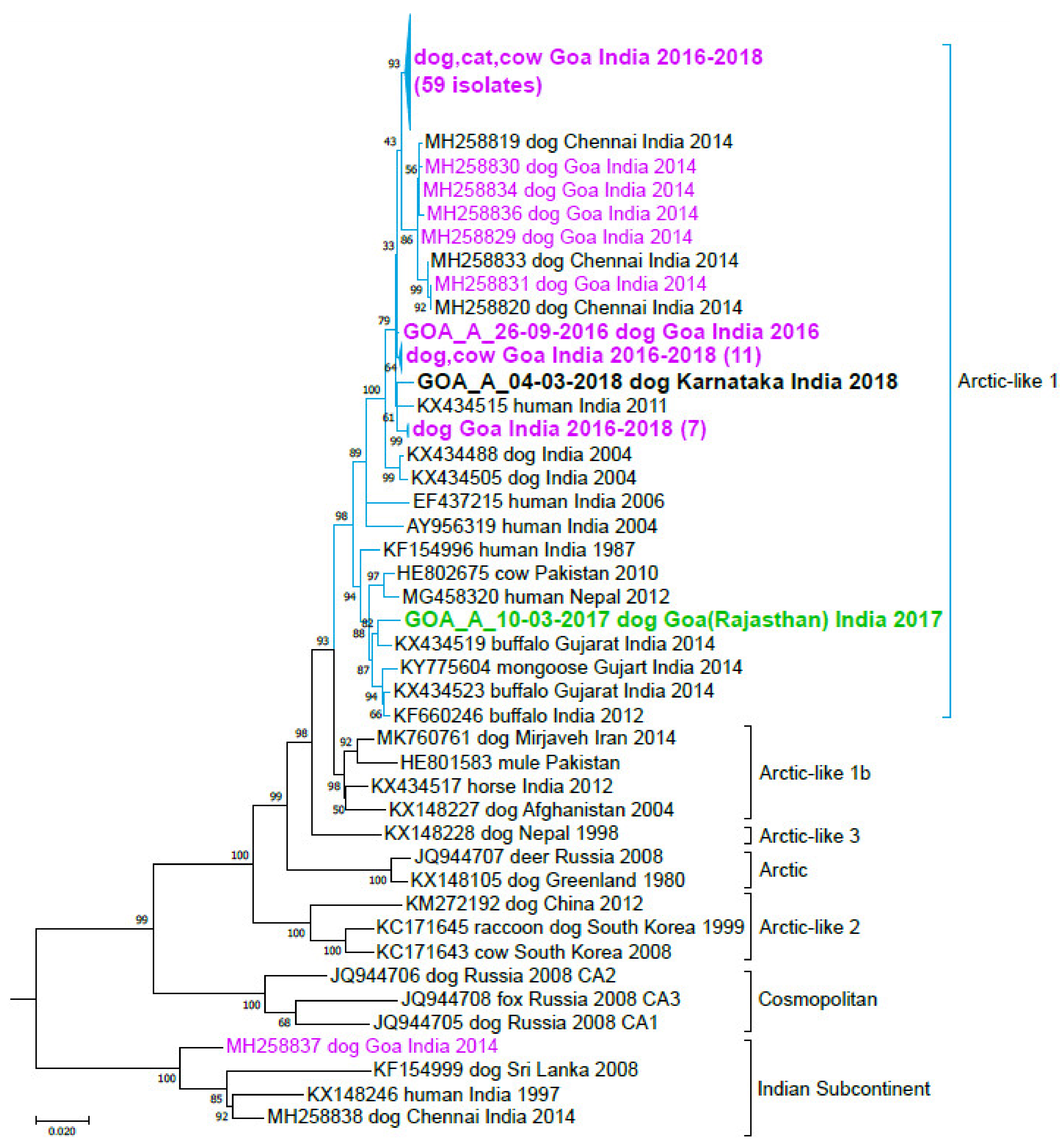
3.3. India
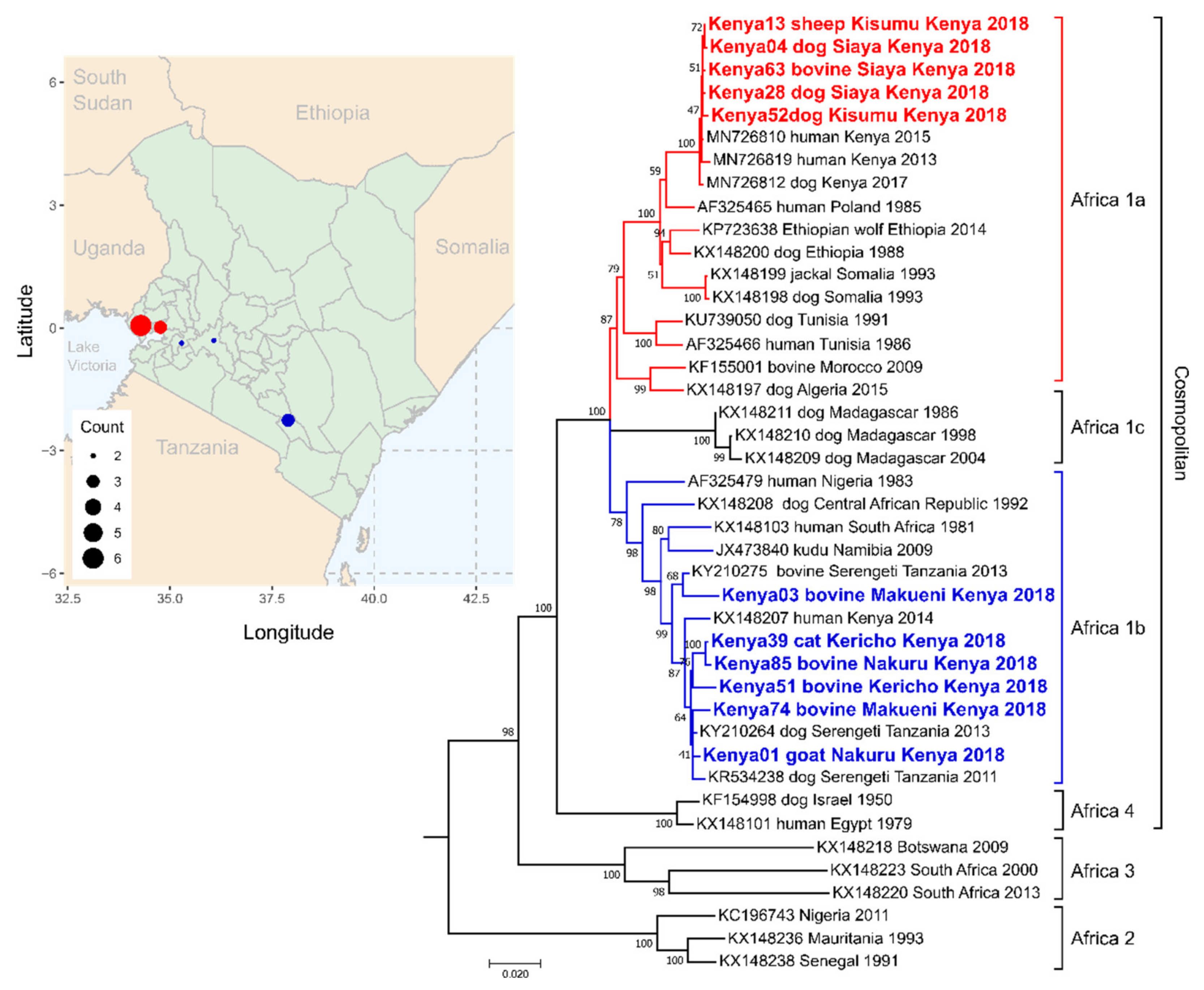
3.4. Kenya
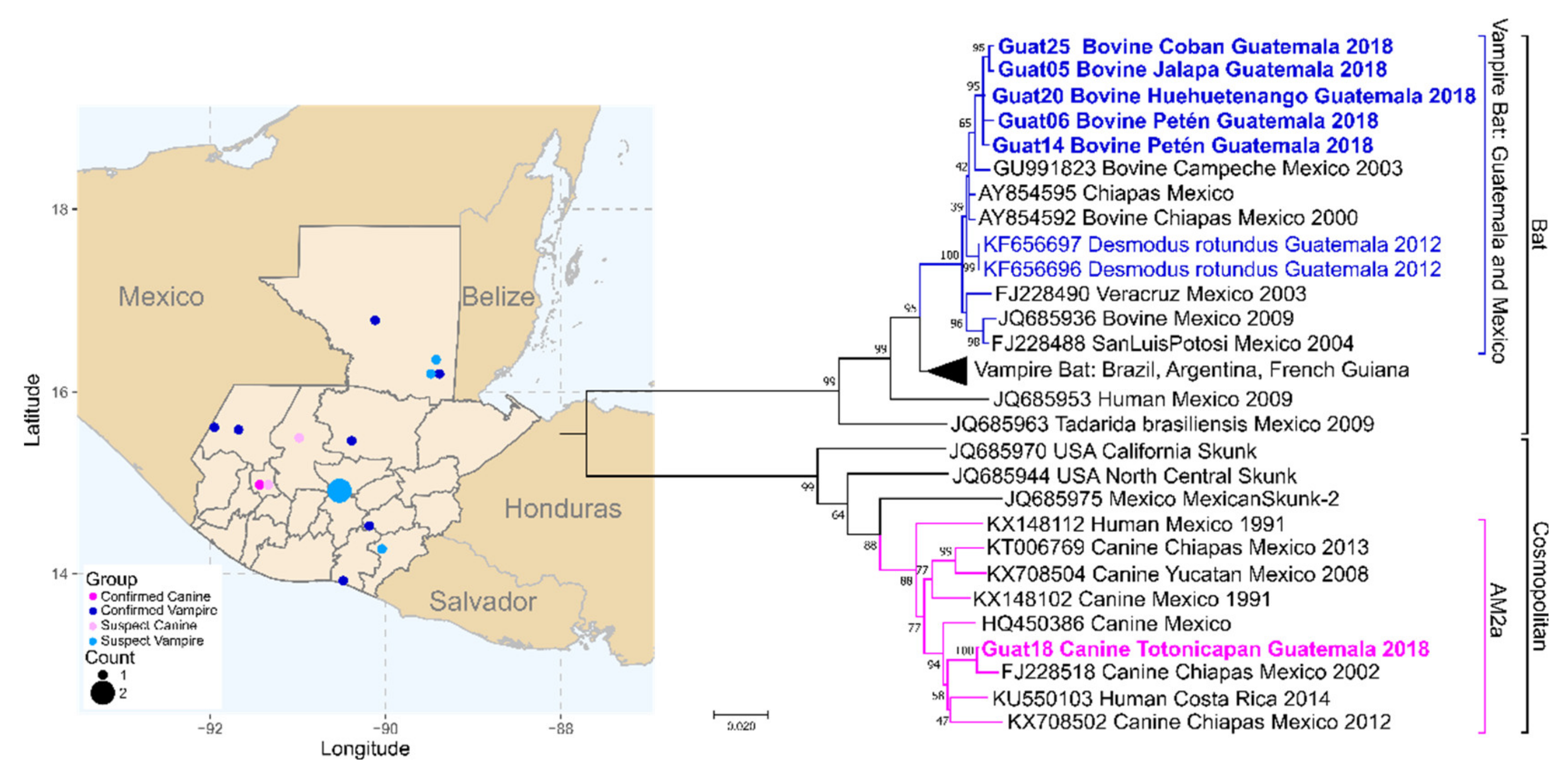
3.5. Guatemala
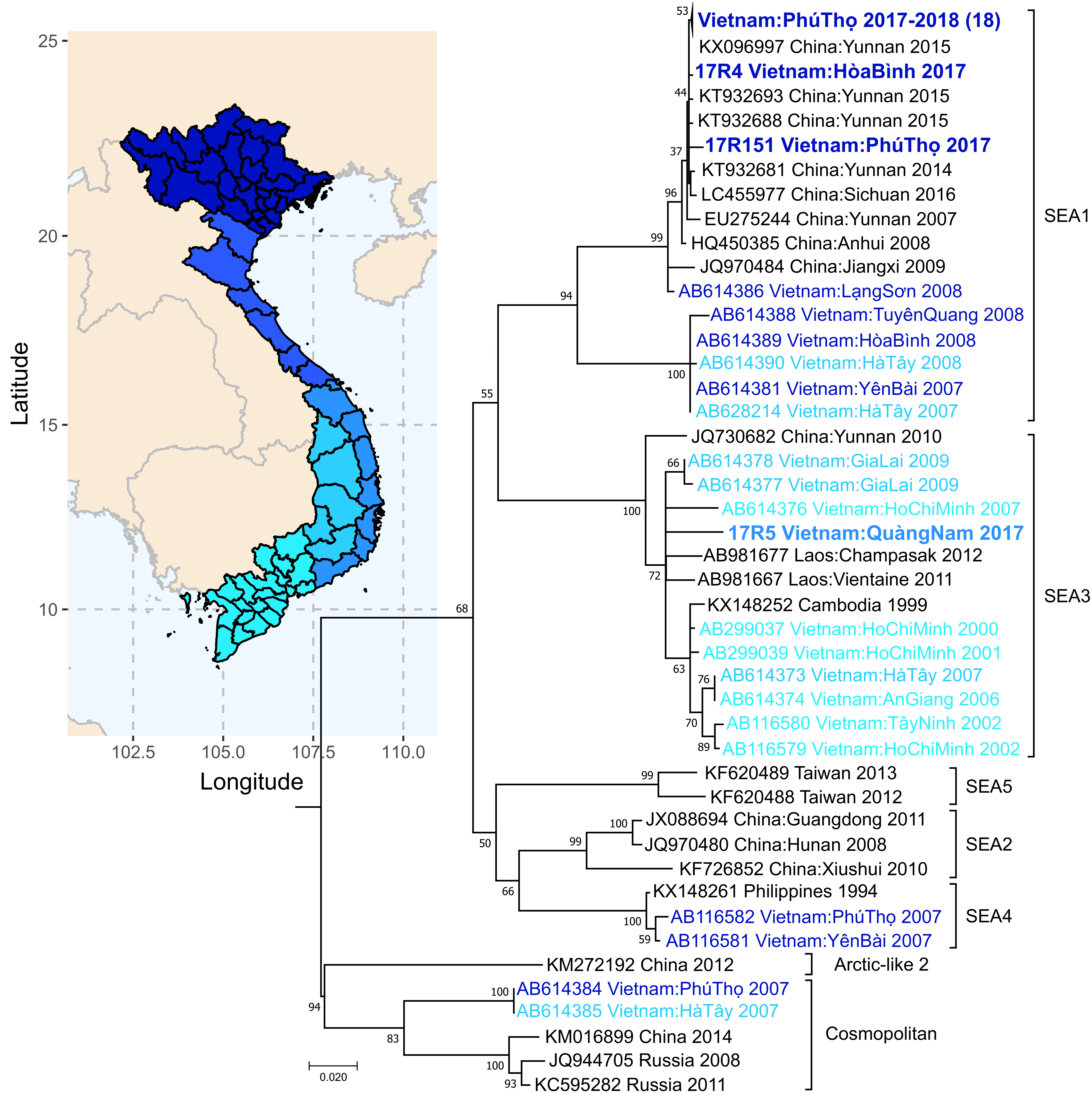
3.6. Vietnam
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Virus Taxonomy: 2019 Release. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 19 October 2020).
- World Health Organization. WHO Expert Consultation on Rabies: Third Report; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Hampson, K.; Coudeville, L.; Lembo, T.; Sambo, M.; Kieffer, A.; Attlan, M.; Barrat, J.; Blanton, J.D.; Briggs, D.J.; Cleaveland, S.; et al. Global Alliance for rabies control partners for rabies. Estimating the global burden of endemic canine rabies. PLoS Negl. Trop. Dis. 2015, 9, e0003709. [Google Scholar]
- Hampson, K.; Dushoff, J.; Bingham, J.; Bruckner, G.; Ali, Y.H.; Dobson, A. Synchronous cycles of domestic dog rabies in sub-Saharan Africa and the impact of control efforts. Proc. Natl. Acad. Sci. USA 2007, 104, 7717–7722. [Google Scholar] [CrossRef] [PubMed]
- Belotto, A.; Leanes, L.F.; Schneider, M.C.; Tamayo, H.; Correa, E. Overview of rabies in the Americas. Virus Res. 2005, 111, 5–12. [Google Scholar] [CrossRef] [PubMed]
- Coleman, P.G.; Dye, C. Immunization coverage required to prevent outbreaks of dog rabies. Vaccine 1996, 14, 185–186. [Google Scholar] [CrossRef]
- Cleaveland, S.; Kaare, M.; Tiringa, P.; Mlengeya, T.; Barrat, J. A dog rabies vaccination campaign in rural Africa: Impact on the incidence of dog rabies and human dog-bite injuries. Vaccine 2003, 21, 1965–1973. [Google Scholar] [CrossRef]
- Tierkel, E.S.; Graves, L.M.; Tuggle, H.G.; Wadley, S.L. Effective control of an outbreak of rabies in Memphis and Shelby County, Tennessee. Am. J. Public Health Nat. Health 1950, 40, 1084–1088. [Google Scholar] [CrossRef]
- Wallace, R.M.; Undurraga, E.A.; Blanton, J.D.; Cleaton, J.; Franka, R. Elimination of dog-mediated human rabies deaths by 2030: Needs assessment and alternatives for progress based on dog vaccination. Front. Vet. Sci. 2017, 4, 9. [Google Scholar] [CrossRef]
- World Health Organization. Zero by 30: The Global Strategic Plan to End Human Deaths from Dog-Mediated Rabies by 2030; Abela-Ridder, B., Ed.; Neglected Zoonotic Diseases; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Lembo, T.; Hampson, K.; Kaare, M.T.; Ernest, E.; Knobel, D.; Kazwala, R.R.; Haydon, D.T.; Cleaveland, S. The feasibility of canine rabies elimination in Africa: Dispelling doubts with data. PLoS Negl. Trop. Dis. 2010, 4, e626. [Google Scholar] [CrossRef]
- Kitala, P.M.; McDermott, J.J.; Kyule, M.N.; Gathuma, J.M. Community-based active surveillance for rabies in Machakos District, Kenya. Prev. Vet. Med. 2000, 44, 73–85. [Google Scholar] [CrossRef]
- Cleaveland, S.; Fèvre, E.M.; Kaare, M.; Coleman, P.G. Estimating human rabies mortality in the United Republic of Tanzania from dog bite injuries. Bull. World Heal. Organ. 2002, 80, 304–310. [Google Scholar]
- Taylor, L.H.; Hampson, K.; Fahrion, A.; Abela-Ridder, B.; Nel, L.H. Difficulties in estimating the human burden of canine rabies. Acta Tropica 2017, 165, 133–140. [Google Scholar] [CrossRef]
- Deressa, A.; Ali, A.; Bayene, M.; Selassie, B.N.; Yimer, E.; Hussen, K. The status of rabies in Ethiopia: A retrospective record review. Ethiop. J. Health Dev. 2010, 24, 127–132. [Google Scholar] [CrossRef]
- Ly, S.; Buchy, P.; Heng, N.Y.; Ong, S.; Chhor, N.; Bourhy, H.; Vong, S. Rabies situation in Cambodia. PLoS Negl. Trop. Dis. 2009, 3, e511. [Google Scholar] [CrossRef]
- Knobel, D.L.; Cleaveland, S.; Coleman, P.G.; Fevre, E.M.; Meltzer, M.I.; Miranda, M.E.; Shaw, A.; Zinsstag, J.; Meslin, F.X. Re-evaluating the burden of rabies in Africa and Asia. Bull. World Health Organ. 2005, 83, 360–368. [Google Scholar] [PubMed]
- Banyard, A.C.; Horton, D.L.; Freuling, C.; Muller, T.; Fooks, A.R. Control and prevention of canine rabies: The need for building laboratory-based surveillance capacity. Antivir. Res. 2013, 98, 357–364. [Google Scholar] [CrossRef]
- Townsend, S.E.; Lembo, T.; Cleaveland, S.; Meslin, F.X.; Miranda, M.E.; Putra, A.A.; Haydon, D.T.; Hampson, K. Surveillance guidelines for disease elimination: A case study of canine rabies. Comp. Immunol. Microbiol. Infect. Dis. 2013, 36, 249–261. [Google Scholar] [CrossRef]
- World Organization of Animal Health (OIE). Rabies (infection with rabies virus and other lyssaviruses). In Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, 8th ed.; World Organization of Animal Health: Paris, France, 2018. [Google Scholar]
- Franka, R.; Wallace, R. Rabies diagnosis and surveillance in animals in the era of rabies elimination. Rev. Sci. Tech. 2018, 37, 359–370. [Google Scholar] [CrossRef]
- Wu, X.; Hu, R.; Zhang, Y.; Dong, G.; Rupprecht, C.E. Reemerging rabies and lack of systemic surveillance in People’s of China. Emerg. Infect. Dis. 2009, 15, 1159–1164. [Google Scholar] [CrossRef]
- Zhang, J.; Jin, Z.; Sun, G.Q.; Zhou, T.; Ruan, S. Analysis of rabies in China: Transmission dynamics and control. PLoS ONE 2011, 6, e20891. [Google Scholar] [CrossRef] [PubMed]
- Lapiz, S.M.; Miranda, M.E.; Garcia, R.G.; Daguro, L.I.; Paman, M.D.; Madrinan, F.P.; Rances, P.A.; Briggs, D.J. Implementation of an intersectoral program to eliminate human and canine rabies: The bohol rabies prevention and elimination project. PLoS Negl. Trop. Dis. 2012, 6, e1891. [Google Scholar] [CrossRef]
- Meslin, F.X.; Briggs, D.J. Eliminating canine rabies, the principal source of human infection: What will it take? Antivir. Res. 2013, 98, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Badrane, H.; Tordo, N. Host switching in Lyssavirus history from the Chiroptera to the Carnivora orders. J. Virol. 2001, 75, 8096–8104. [Google Scholar] [CrossRef] [PubMed]
- Streicker, D.G.; Turmelle, A.S.; Vonhof, M.J.; Kuzmin, I.V.; McCracken, G.F.; Rupprecht, C.E. Host phylogeny constrains cross-species emergence and establishment of rabies virus in bats. Science 2010, 329, 676–679. [Google Scholar] [CrossRef]
- Bourhy, H.; Kissi, B.; Audry, L.; Smreczak, M.; Sadkowska-Todys, M.; Kulonen, K.; Tordo, N.; Zmudzinski, J.F.; Holmes, E.C. Ecology and evolution of rabies virus in Europe. J. Gen. Virol. 1999, 80, 2545–2557. [Google Scholar] [CrossRef]
- Rupprecht, C.E.; Hanlon, C.A.; Hemachudha, T. Rabies re-examined. Lancet Infect. Dis. 2002, 2, 327–343. [Google Scholar] [CrossRef]
- Mollentze, N.; Biek, R.; Streicker, D.G. The role of viral evolution in rabies host shifts and emergence. Curr. Opin. Virol. 2014, 8, 68–72. [Google Scholar] [CrossRef]
- Troupin, C.; Dacheux, L.; Tanguy, M.; Sabeta, C.; Blanc, H.; Bouchier, C.; Vignuzzi, M.; Duchene, S.; Holmes, E.C.; Bourhy, H.; et al. Large-Scale phylogenomic analysis reveals the complex evolutionary history of rabies virus in multiple carnivore hosts. PLoS Pathog. 2016, 12, e1006041. [Google Scholar] [CrossRef]
- Bourhy, H.; Reynes, J.M.; Dunham, E.J.; Dacheux, L.; Larrous, F.; Huong, V.T.; Xu, G.; Yan, J.; Miranda, M.E.; Holmes, E.C.; et al. The origin and phylogeography of dog rabies virus. J. Gen. Virol. 2008, 89, 2673–2681. [Google Scholar] [CrossRef]
- Zieger, U.; Marston, D.A.; Sharma, R.; Chikweto, A.; Tiwari, K.; Sayyid, M.; Louison, B.; Goharriz, H.; Voller, K.; Breed, A.C.; et al. The phylogeography of rabies in Grenada, West Indies, and implications for control. PLoS Negl. Trop. Dis. 2014, 8, e3251. [Google Scholar] [CrossRef]
- Nadin-Davis, S.A. Molecular epidemiology. In Rabies; Jackson, A.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 123–177. [Google Scholar]
- Velasco-Villa, A.; Orciari, L.A.; Juarez-Islas, V.; Gomez-Sierra, M.; Padilla-Medina, I.; Flisser, A.; Souza, V.; Castillo, A.; Franka, R.; Escalante-Mane, M.; et al. Molecular diversity of rabies viruses associated with bats in Mexico and other countries of the Americas. J. Clin. Microbiol. 2006, 44, 1697–1710. [Google Scholar] [CrossRef]
- Nadin-Davis, S.A.; Huang, W.; Armstrong, J.; Casey, G.A.; Bahloul, C.; Tordo, N.; Wandeler, A.I. Antigenic and genetic divergence of rabies viruses from bat species indigenous to Canada. Virus Res. 2001, 74, 139–156. [Google Scholar] [CrossRef]
- Kuzmin, I.V.; Shi, M.; Orciari, L.A.; Yager, P.A.; Velasco-Villa, A.; Kuzmina, N.A.; Streicker, D.G.; Bergman, D.L.; Rupprecht, C.E. Molecular inferences suggest multiple host shifts of rabies viruses from bats to mesocarnivores in Arizona during 2001–2009. PLoS Pathog. 2012, 8, e1002786. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.S.; Orciari, L.A.; Yager, P.A.; Seidel, H.D.; Warner, C.K. Epidemiologic and historical relationships among 87 rabies virus isolates as determined by limited sequence analysis. J. Infect. Dis. 1992, 166, 296–307. [Google Scholar] [CrossRef]
- Wiktor, T.J.; Koprowski, H. Antigenic variants of rabies virus. J. Exp. Med. 1980, 152, 99–112. [Google Scholar] [CrossRef]
- De Mattos, C.C.; De Mattos, C.A.; Loza-Rubio, E.; Aguilar-Setien, A.; Orciari, L.A.; Smith, J.S. Molecular characterization of rabies virus isolates from Mexico: Implications for transmission dynamics and human risk. Am. J. Trop. Med. Hyg. 1999, 61, 587–597. [Google Scholar] [CrossRef]
- Ma, X.; Monroe, B.P.; Cleaton, J.M.; Orciari, L.A.; Gigante, C.M.; Kirby, J.D.; Chipman, R.B.; Fehlner-Gardiner, C.; Gutierrez Cedillo, V.; Petersen, B.W.; et al. Public veterinary medicine: Public health: Rabies surveillance in the US during 2018. J. Am. Vet. Med. Assoc. 2020, 256, 195–208. [Google Scholar] [CrossRef] [PubMed]
- Pant, G.R.; Lavenir, R.; Wong, F.Y.; Certoma, A.; Larrous, F.; Bhatta, D.R.; Bourhy, H.; Stevens, V.; Dacheux, L. Recent emergence and spread of an Arctic-related phylogenetic lineage of rabies virus in Nepal. PLoS Negl. Trop. Dis. 2013, 7, e2560. [Google Scholar] [CrossRef]
- Steck, F.; Wandeler, A. The epidemiology of fox rabies in Europe. Epidemiol. Rev. 1980, 2, 71–96. [Google Scholar] [CrossRef] [PubMed]
- Horton, D.L.; McElhinney, L.M.; Freuling, C.M.; Marston, D.A.; Banyard, A.C.; Goharrriz, H.; Wise, E.; Breed, A.C.; Saturday, G.; Kolodziejek, J.; et al. Complex epidemiology of a zoonotic disease in a culturally diverse region: Phylogeography of rabies virus in the Middle East. PLoS Negl. Trop. Dis. 2015, 9, e0003569. [Google Scholar] [CrossRef]
- Zhang, Y.; Vrancken, B.; Feng, Y.; Dellicour, S.; Yang, Q.; Yang, W.; Zhang, Y.; Dong, L.; Pybus, O.G.; Zhang, H.; et al. Cross-border spread, lineage displacement and evolutionary rate estimation of rabies virus in Yunnan Province, China. Virol. J. 2017, 14, 102. [Google Scholar] [CrossRef]
- Brunker, K.; Marston, D.A.; Horton, D.L.; Cleaveland, S.; Fooks, A.R.; Kazwala, R.; Ngeleja, C.; Lembo, T.; Sambo, M.; Mtema, Z.J.; et al. Elucidating the phylodynamics of endemic rabies virus in eastern Africa using whole-genome sequencing. Virus Evol. 2015, 1, vev011. [Google Scholar] [CrossRef]
- Tordo, N.; Kouknetzoff, A. The rabies virus genome: An overview. Onderstepoort J. Vet. Res. 1993, 60, 263–269. [Google Scholar]
- Dietzschold, B.; Wunner, W.H.; Wiktor, T.J.; Lopes, A.D.; Lafon, M.; Smith, C.L.; Koprowski, H. Characterization of an antigenic determinant of the glycoprotein that correlates with pathogenicity of rabies virus. Proc. Natl. Acad. Sci. USA 1983, 80, 70–74. [Google Scholar] [CrossRef]
- Dietzschold, B.; Li, J.; Faber, M.; Schnell, M. Concepts in the pathogenesis of rabies. Future Virol. 2008, 3, 481–490. [Google Scholar] [CrossRef]
- Cox, J.H.; Dietzschold, B.; Schneider, L.G. Rabies virus glycoprotein. II. Biological and serological characterization. Infect. Immun. 1977, 16, 754–759. [Google Scholar] [CrossRef]
- Koboldt, D.C.; Steinberg, K.M.; Larson, D.E.; Wilson, R.K.; Mardis, E.R. The next-generation sequencing revolution and its impact on genomics. Cell 2013, 155, 27–38. [Google Scholar] [CrossRef]
- Muir, P.; Li, S.; Lou, S.; Wang, D.; Spakowicz, D.J.; Salichos, L.; Zhang, J.; Weinstock, G.M.; Isaacs, F.; Rozowsky, J.; et al. The real cost of sequencing: Scaling computation to keep pace with data generation. Genome Biol. 2016, 17, 53. [Google Scholar] [CrossRef]
- Biesecker, L.G.; Burke, W.; Kohane, I.; Plon, S.E.; Zimmern, R. Next-generation sequencing in the clinic: Are we ready? Nat. Rev. Genet. 2012, 13, 818–824. [Google Scholar] [CrossRef][Green Version]
- Sboner, A.; Mu, X.J.; Greenbaum, D.; Auerbach, R.K.; Gerstein, M.B. The real cost of sequencing: Higher than you think! Genome Biol. 2011, 12, 125. [Google Scholar] [CrossRef]
- Fooks, A.R.; Johnson, N.; Freuling, C.M.; Wakeley, P.R.; Banyard, A.C.; McElhinney, L.M.; Marston, D.A.; Dastjerdi, A.; Wright, E.; Weiss, R.A.; et al. Emerging technologies for the detection of rabies virus: Challenges and hopes in the 21st century. PLoS Negl. Trop. Dis. 2009, 3, e530. [Google Scholar] [CrossRef] [PubMed]
- Picard-Meyer, E.; Barrat, J.; Cliquet, F. Use of filter paper (FTA) technology for sampling, recovery and molecular characterisation of rabies viruses. J. Virol. Methods 2007, 140, 174–182. [Google Scholar] [CrossRef]
- Hofmann, M.A.; Thur, B.; Liu, L.; Gerber, M.; Stettler, P.; Moser, C.; Bossy, S. Rescue of infectious classical swine fever and foot-and-mouth disease virus by RNA transfection and virus detection by RT-PCR after extended storage of samples in Trizol. J. Virol. Methods 2000, 87, 29–39. [Google Scholar] [CrossRef]
- Lou, J.J.; Mirsadraei, L.; Sanchez, D.E.; Wilson, R.W.; Shabihkhani, M.; Lucey, G.M.; Wei, B.; Singer, E.J.; Mareninov, S.; Yong, W.H.; et al. A review of room temperature storage of biospecimen tissue and nucleic acids for anatomic pathology laboratories and biorepositories. Clin. Biochem. 2014, 47, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Chin, C.D.; Linder, V.; Sia, S.K. Commercialization of microfluidic point-of-care diagnostic devices. Lab Chip 2012, 12, 2118–2134. [Google Scholar] [CrossRef]
- Bissonnette, L.; Bergeron, M.G. Portable devices and mobile instruments for infectious diseases point-of-care testing. Expert Rev. Mol. Diagn. 2017, 17, 471–494. [Google Scholar] [CrossRef]
- Holland, C.A.; Kiechle, F.L. Point-of-care molecular diagnostic systems—Past, present and future. Curr. Opin. Microbiol. 2005, 8, 504–509. [Google Scholar] [CrossRef]
- Quick, J.; Loman, N.J.; Duraffour, S.; Simpson, J.T.; Severi, E.; Cowley, L.; Bore, J.A.; Koundouno, R.; Dudas, G.; Mikhail, A.; et al. Real-time, portable genome sequencing for Ebola surveillance. Nature 2016, 530, 228–232. [Google Scholar] [CrossRef]
- Faria, N.R.; Azevedo, R.; Kraemer, M.U.G.; Souza, R.; Cunha, M.S.; Hill, S.C.; Theze, J.; Bonsall, M.B.; Bowden, T.A.; Rissanen, I.; et al. Zika virus in the Americas: Early epidemiological and genetic findings. Science 2016, 352, 345–349. [Google Scholar] [CrossRef]
- World Health Organization. The direct fluorescent antibody test. In Laboratory Techniques in Rabies, 5th ed.; Rupprecht, C.E., Fooks, A.R., Abela-Ridder, B., Eds.; World Health Organization: Geneva, Switzerland, 2019; Volume 1. [Google Scholar]
- Gigante, C.M.; Dettinger, L.; Powell, J.W.; Seiders, M.; Condori, R.E.C.; Griesser, R.; Okogi, K.; Carlos, M.; Pesko, K.; Breckenridge, M.; et al. Multi-site evaluation of the LN34 pan-lyssavirus real-time RT-PCR assay for post-mortem rabies diagnostics. PLoS ONE 2018, 13, e0197074. [Google Scholar] [CrossRef]
- Wadhwa, A.; Wilkins, K.; Gao, J.; Condori Condori, R.E.; Gigante, C.M.; Zhao, H.; Ma, X.; Ellison, J.A.; Greenberg, L.; Velasco-Villa, A.; et al. A Pan-lyssavirus taqman real-time RT-PCR Assay for the detection of highly variable rabies virus and other lyssaviruses. PLoS Negl. Trop. Dis. 2017, 11, e0005258. [Google Scholar] [CrossRef]
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A cross-platform and ultrafast toolkit for FASTA/Q file manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Condori-Condori, R.E.; Streicker, D.G.; Cabezas-Sanchez, C.; Velasco-Villa, A. Enzootic and epizootic rabies associated with vampire bats, Peru. Emerg. Infect. Dis. 2013, 19, 1463–1469. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.A. BioEdit: A User-Friendly Biological Sequence Alignment Editor and Analysis Program for Windows 95/98/NT; Nucleic Acids Symposium Series; Oxford University Press: Oxford, UK, 1999; pp. 95–98. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Rstudio Team. RStudio: Integrated Development for R; RStudio, Inc: Boston, MA, USA, 2015. [Google Scholar]
- R Core Team, R. A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2016. [Google Scholar]
- Hijmans, R.J. Raster: Geographic Data Analysis and Modeling. Available online: https://CRAN.R-project.org/package=raster (accessed on 12 February 2020).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer-Verlag: New York, NY, USA, 2009. [Google Scholar]
- Jain, M.; Tyson, J.R.; Loose, M.; Ip, C.L.C.; Eccles, D.A.; O’Grady, J.; Malla, S.; Leggett, R.M.; Wallerman, O.; Jansen, H.J.; et al. MinION analysis and reference consortium: Phase 2 data release and analysis of R9.0 chemistry. F1000Research 2017, 6, 760. [Google Scholar] [CrossRef] [PubMed]
- Tyson, J.R.; O’Neil, N.J.; Jain, M.; Olsen, H.E.; Hieter, P.; Snutch, T.P. MinION-based long-read sequencing and assembly extends the Caenorhabditis elegans reference genome. Genome Res. 2018, 28, 266–274. [Google Scholar] [CrossRef]
- Cretu Stancu, M.; Van Roosmalen, M.J.; Renkens, I.; Nieboer, M.M.; Middelkamp, S.; De Ligt, J.; Pregno, G.; Giachino, D.; Mandrile, G.; Espejo Valle-Inclan, J.; et al. Mapping and phasing of structural variation in patient genomes using nanopore sequencing. Nat. Commun. 2017, 8, 1326. [Google Scholar] [CrossRef]
- Wick, R.R.; Judd, L.M.; Holt, K.E. Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 2019, 20, 129. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338–345. [Google Scholar] [CrossRef]
- Loman, N.J.; Quick, J.; Simpson, J.T. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods 2015, 12, 733–735. [Google Scholar] [CrossRef]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef] [PubMed]
- Solomon, T.; Marston, D.; Mallewa, M.; Felton, T.; Shaw, S.; McElhinney, L.M.; Das, K.; Mansfield, K.; Wainwright, J.; Kwong, G.N.; et al. Paralytic rabies after a two week holiday in India. BMJ 2005, 331, 501–503. [Google Scholar] [CrossRef]
- Deventhiran, J.; Srinivasan, B.; Seeralan, M.S.; Kanagaraj, V.; Raju, S.; Kathaperumal, K.; Nissly, R.; Kasibhatla, S.M.; Kale, M.M.; Kulkarni-Kale, U.; et al. Continued circulation, recombination and evolution of the ancient subcontinent lineage despite predominance of the recent arctic-like lineage rabies viruses (RABV) in India. BioRxiv 2018. BioRxiv:398180. [Google Scholar]
- Kissi, B.; Tordo, N.; Bourhy, H. Genetic polymorphism in the rabies virus nucleoprotein gene. Virology 1995, 209, 526–537. [Google Scholar] [CrossRef]
- Nadin-Davis, S.A.; Casey, G.A.; Wandeler, A.I. A molecular epidemiological study of rabies virus in central Ontario and western Quebec. J. Gen. Virol. 1994, 75, 2575–2583. [Google Scholar] [CrossRef]
- Yamagata, J.; Ahmed, K.; Khawplod, P.; Mannen, K.; Xuyen, D.K.; Loi, H.H.; Dung, N.V.; Nishizono, A. Molecular epidemiology of rabies in Vietnam. Microbiol. Immunol. 2007, 51, 833–840. [Google Scholar] [CrossRef]
- Ellison, J.A.; Gilbert, A.T.; Recuenco, S.; Moran, D.; Alvarez, D.A.; Kuzmina, N.; Garcia, D.L.; Peruski, L.F.; Mendonca, M.T.; Lindblade, K.A.; et al. Bat rabies in Guatemala. PLoS Negl. Trop. Dis. 2014, 8, e3070. [Google Scholar] [CrossRef]
- Nadin-Davis, S.A.; Colville, A.; Trewby, H.; Biek, R.; Real, L. Application of high-throughput sequencing to whole rabies viral genome characterisation and its use for phylogenetic re-evaluation of a raccoon strain incursion into the province of Ontario. Virus Res. 2017, 232, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Trewby, H.; Nadin-Davis, S.A.; Real, L.A.; Biek, R. Processes underlying rabies virus incursions across US-Canada border as revealed by whole-genome phylogeography. Emerg. Infect. Dis. 2017, 23, 1454–1461. [Google Scholar] [CrossRef]
- Hanke, D.; Freuling, C.M.; Fischer, S.; Hueffer, K.; Hundertmark, K.; Nadin-Davis, S.; Marston, D.; Fooks, A.R.; Botner, A.; Mettenleiter, T.C.; et al. Spatio-temporal analysis of the genetic diversity of Arctic rabies viruses and their reservoir hosts in Greenland. PLoS Negl. Trop. Dis. 2016, 10, e0004779. [Google Scholar] [CrossRef] [PubMed]
- Brunker, K.; Jaswant, G.; Thumbi, S.; Lushasi, K.; Lugelo, A.; Czupryna, A.M.; Ade, F.; Wambura, G.; Chuchu, V.; Steenson, R.; et al. Rapid in-country sequencing of whole virus genomes to inform rabies elimination programmes. Wellcome Open Res. 2020, 5. [Google Scholar] [CrossRef]
- Sudarshan, M.K.; Madhusudana, S.N.; Mahendra, B.J.; Rao, N.S.; Ashwath Narayana, D.H.; Abdul Rahman, S.; Meslin, F.; Lobo, D.; Ravikumar, K. Gangaboraiah, assessing the burden of human rabies in India: Results of a national multi-center epidemiological survey. Int. J. Infect. Dis. 2007, 11, 29–35. [Google Scholar] [CrossRef]
- Reddy, R.V.; Mohana Subramanian, B.; Surendra, K.S.; Babu, R.P.; Rana, S.K.; Manjari, K.S.; Srinivasan, V.A. Rabies virus isolates of India—Simultaneous existence of two distinct evolutionary lineages. Infect. Genet. Evol. 2014, 27, 163–172. [Google Scholar] [CrossRef]
- Nadin-Davis, S.A.; Turner, G.; Paul, J.P.; Madhusudana, S.N.; Wandeler, A.I. Emergence of Arctic-like rabies lineage in India. Emerg. Infect. Dis. 2007, 13, 111–116. [Google Scholar] [CrossRef]
- Nagarajan, T.; Mohanasubramanian, B.; Seshagiri, E.V.; Nagendrakumar, S.B.; Saseendranath, M.R.; Satyanarayana, M.L.; Thiagarajan, D.; Rangarajan, P.N.; Srinivasan, V.A. Molecular epidemiology of rabies virus isolates in India. J. Clin. Microbiol. 2006, 44, 3218–3224. [Google Scholar] [CrossRef]
- Nagarajan, T.; Nagendrakumar, S.B.; Mohanasubramanian, B.; Rajalakshmi, S.; Hanumantha, N.R.; Ramya, R.; Thiagarajan, D.; Srinivasan, V.A. Phylogenetic analysis of nucleoprotein gene of dog rabies virus isolates from Southern India. Infect. Genet. Evol. 2009, 9, 976–982. [Google Scholar] [CrossRef]
- Manjunatha Reddy, G.B.; Krishnappa, S.; Vinayagamurthy, B.; Singh, R.; Singh, K.P.; Saminathan, M.; Sajjanar, B.; Rahman, H. Molecular epidemiology of rabies virus circulating in domestic animals in India. Virusdisease 2018, 29, 362–368. [Google Scholar] [CrossRef] [PubMed]
- Sudarshan, M.K.; Mahendra, B.J.; Narayan, D.H. A community survey of dog bites, anti-rabies treatment, rabies and dog population management in Bangalore city. J. Commun. Dis. 2001, 33, 245–251. [Google Scholar] [PubMed]
- Reece, J.F.; Chawla, S.K. Control of rabies in Jaipur, India, by the sterilisation and vaccination of neighbourhood dogs. Vet. Rec. 2006, 159, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Abbas, S.S.; Venkataramanan, V.; Pathak, G.; Kakkar, M. Roadmap to combat zoonoses in India. Rabies control initiative in Tamil Nadu, India: A test case for the ’One Health’ approach. Int. Health 2011, 3, 231–239. [Google Scholar] [CrossRef]
- Totton, S.C.; Wandeler, A.I.; Zinsstag, J.; Bauch, C.T.; Ribble, C.S.; Rosatte, R.C.; McEwen, S.A. Stray dog population demographics in Jodhpur, India following a population control/rabies vaccination program. Prev. Vet. Med. 2010, 97, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Gibson, A.D.; Mazeri, S.; Lohr, F.; Mayer, D.; Burdon Bailey, J.L.; Wallace, R.M.; Handel, I.G.; Shervell, K.; Bronsvoort, B.M.D.; Mellanby, R.J.; et al. One million dog vaccinations recorded on mHealth innovation used to direct teams in numerous rabies control campaigns. PLoS ONE 2018, 13, e0200942. [Google Scholar] [CrossRef]
- Gibson, A.D.; Ohal, P.; Shervell, K.; Handel, I.G.; Bronsvoort, B.M.; Mellanby, R.J.; Gamble, L. Vaccinate-assess-move method of mass canine rabies vaccination utilising mobile technology data collection in Ranchi, India. BMC Infect. Dis. 2015, 15, 589. [Google Scholar] [CrossRef]
- Johnson, N.; McElhinney, L.M.; Ali, Y.H.; Saeed, I.K.; Fooks, A.R. Molecular epidemiology of canid rabies in Sudan: Evidence for a common origin of rabies with Ethiopia. Virus Res. 2004, 104, 201–205. [Google Scholar] [CrossRef]
- Schneider, M.C.; Belotto, A.; Ade, M.P.; Leanes, L.F.; Correa, E.; Tamayo, H.; Medina, G.; Rodrigues, M.J. Epidemiologic situation of human rabies in Latin America in 2004. Epidemiol. Bull. 2005, 26, 2–4. [Google Scholar] [PubMed]
- Lee, D.N.; Papes, M.; Van den Bussche, R.A. Present and potential future distribution of common vampire bats in the Americas and the associated risk to cattle. PLoS ONE 2012, 7, e42466. [Google Scholar] [CrossRef]
- Mayen, F. Haematophagous bats in Brazil, their role in rabies transmission, impact on public health, livestock industry and alternatives to an indiscriminate reduction of bat population. J. Vet. Med. B Infect. Dis. Vet. Public Health 2003, 50, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Putra, A.A.; Hampson, K.; Girardi, J.; Hiby, E.; Knobel, D.; Mardiana, I.W.; Townsend, S.; Scott-Orr, H. Response to a rabies epidemic, Bali, Indonesia, 2008–2011. Emerg. Infect. Dis. 2013, 19, 648–651. [Google Scholar] [CrossRef]
- Windiyaningsih, C.; Wilde, H.; Meslin, F.X.; Suroso, T.; Widarso, H.S. The rabies epidemic on Flores Island, Indonesia (1998–2003). J. Med. Assoc. Thai. 2004, 87, 1389–1393. [Google Scholar]
- Susilawathi, N.M.; Darwinata, A.E.; Dwija, I.B.; Budayanti, N.S.; Wirasandhi, G.A.; Subrata, K.; Susilarini, N.K.; Sudewi, R.A.; Wignall, F.S.; Mahardika, G.N.; et al. Epidemiological and clinical features of human rabies cases in Bali 2008–2010. BMC Infect. Dis. 2012, 12, 81. [Google Scholar] [CrossRef]
- Lardon, Z.; Watier, L.; Brunet, A.; Bernede, C.; Goudal, M.; Dacheux, L.; Rotivel, Y.; Guillemot, D.; Bourhy, H. Imported episodic rabies increases patient demand for and physician delivery of antirabies prophylaxis. PLoS Negl. Trop. Dis. 2010, 4, e723. [Google Scholar] [CrossRef]
- Childs, J.E.; Curns, A.T.; Dey, M.E.; Real, L.A.; Feinstein, L.; Bjornstad, O.N.; Krebs, J.W. Predicting the local dynamics of epizootic rabies among raccoons in the US. Proc. Natl. Acad. Sci. USA 2000, 97, 13666–13671. [Google Scholar] [CrossRef]
- Jeon, S.; Cleaton, J.; Meltzer, M.I.; Kahn, E.B.; Pieracci, E.G.; Blanton, J.D.; Wallace, R. Determining the post-elimination level of vaccination needed to prevent re-establishment of dog rabies. PLoS Negl. Trop. Dis. 2019, 13, e0007869. [Google Scholar] [CrossRef] [PubMed]
- Pieracci, E.G.; Chipman, R.B.; Morgan, C.N.; Brown, C.M.; Kirby, J.D.; Blanton, J.D.; Velasco-Villa, A.; Martin, A.D.; Nelson, K.M.; Singh, A.; et al. Evaluation of rabies virus characterization to enhance early detection of important rabies epizootic events in the US. J. Am. Vet. Med. Assoc. 2020, 256, 66–76. [Google Scholar] [CrossRef]
- Castillo-Neyra, R.; Brown, J.; Borrini, K.; Arevalo, C.; Levy, M.Z.; Buttenheim, A.; Hunter, G.C.; Becerra, V.; Behrman, J.; Paz-Soldan, V.A.; et al. Barriers to dog rabies vaccination during an urban rabies outbreak: Qualitative findings from Arequipa, Peru. PLoS Negl. Trop. Dis. 2017, 11, e0005460. [Google Scholar] [CrossRef]
- Nguyen, A.K.; Nguyen, D.V.; Ngo, G.C.; Nguyen, T.T.; Inoue, S.; Yamada, A.; Dinh, X.K.; Nguyen, D.V.; Phan, T.X.; Pham, B.Q.; et al. Molecular epidemiology of rabies virus in Vietnam (2006–2009). Jpn. J. Infect. Dis. 2011, 64, 391–396. [Google Scholar]
- Guo, Z.; Tao, X.; Yin, C.; Han, N.; Yu, J.; Li, H.; Liu, H.; Fang, W.; Adams, J.; Wang, J.; et al. National borders effectively halt the spread of rabies: The current rabies epidemic in China is dislocated from cases in neighboring countries. PLoS Negl. Trop. Dis. 2013, 7, e2039. [Google Scholar] [CrossRef]
- Zhang, H.L.; Zhang, Y.Z.; Yang, W.H.; Tao, X.Y.; Li, H.; Ding, J.C.; Feng, Y.; Yang, D.J.; Zhang, J.; He, J.; et al. Molecular epidemiology of reemergent rabies in Yunnan Province, southwestern China. Emerg. Infect. Dis. 2014, 20, 1433–1442. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.P.; Zhou, H.; Wu, H.; Tao, X.Y.; Rayner, S.; Wang, S.M.; Tang, Q.; Liang, G.D. Analysis on factors related to rabies epidemic in China from 2007–2011. Virol. Sin. 2012, 27, 132–143. [Google Scholar] [CrossRef]
- Tao, X.Y.; Tang, Q.; Rayner, S.; Guo, Z.Y.; Li, H.; Lang, S.L.; Yin, C.P.; Han, N.; Fang, W.; Adams, J.; et al. Molecular phylodynamic analysis indicates lineage displacement occurred in Chinese rabies epidemics between 1949 to 2010. PLoS Negl. Trop. Dis. 2013, 7, e2294. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer | Sequence |
|---|---|
| NgeneFor | TTTCTGTTGGTGCTGATATTGCAATGTAACACCYCTACAATGGAT |
| NgeneRev | ACTTGCCTGTCGCTCTATCTTCAGGAGGRGTGTTAGTTTTTTTC |
| GgeneFor | TTTCTGTTGGTGCTGATATTGCGATGTGAAAAAAACTATYAACATCCCTC |
| GgeneRev | ACTTGCCTGTCGCTCTATCTTCTGTGAKCTATTGCTTRTGTYCTTCA |
| MinION (Oxford Nanopore Technologies, Oxford, UK) |
| Mic qPCR cycler (Bio Molecular Systems, El Cajon, CA, USA) |
| MiniPCR (Amplyus, Cambridge, MA, USA) |
| E-Gel Electrophoresis System (ThermoFisher, Waltham, MA, USA) |
| Mini Centrifuge (Southern Labware, Cumming, GA, USA) |
| Price/Sample | |||
|---|---|---|---|
| 50 Samples/Run | 20 Samples/Run | 1 Sample/Run | |
| PCR Barcoding | USD 2.11 | USD 2.11 | USD 0.00 |
| Library Preparation | USD 2.79 | USD 6.57 | USD 126.13 |
| Sequencing | USD 10.00 | USD 25.00 | USD 500.00 |
| Total | USD 14.90 | USD 33.68 | USD 626.13 |
| Read Depth | >0 | <50 | >50 | >100 | >1000 | >10,000 |
|---|---|---|---|---|---|---|
| Sequences | 70 | 11 | 58 | 48 | 13 | 5 |
| Raw | 99.842 | 99.213 | 99.958 | 99.968 | 99.965 | 99.958 |
| Polished | 99.933 | 99.778 | 99.961 | 99.965 | 99.987 | 100 |
| Manual | 99.988 | 99.940 | 99.996 | 99.999 | 100 | 100 |
| Country | Samples | Positive | N full | G full | N Partial | G Partial | Percent Success |
|---|---|---|---|---|---|---|---|
| Guatemala | 25 | 13 | 6 | 4 | 0 | 0 | 46.15 |
| India | 104 | 103 | 80 | 97 | 0 | 0 | 93.27 |
| Kenya | 28 | 19 | 10 | 11 | 4 | 5 | 84.21 |
| Vietnam | 21 | 21 | 21 | 21 | 0 | 0 | 100.00 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gigante, C.M.; Yale, G.; Condori, R.E.; Costa, N.C.; Long, N.V.; Minh, P.Q.; Chuong, V.D.; Tho, N.D.; Thanh, N.T.; Thin, N.X.; et al. Portable Rabies Virus Sequencing in Canine Rabies Endemic Countries Using the Oxford Nanopore MinION. Viruses 2020, 12, 1255. https://doi.org/10.3390/v12111255
Gigante CM, Yale G, Condori RE, Costa NC, Long NV, Minh PQ, Chuong VD, Tho ND, Thanh NT, Thin NX, et al. Portable Rabies Virus Sequencing in Canine Rabies Endemic Countries Using the Oxford Nanopore MinION. Viruses. 2020; 12(11):1255. https://doi.org/10.3390/v12111255
Chicago/Turabian StyleGigante, Crystal M., Gowri Yale, Rene Edgar Condori, Niceta Cunha Costa, Nguyen Van Long, Phan Quang Minh, Vo Dinh Chuong, Nguyen Dang Tho, Nguyen Tat Thanh, Nguyen Xuan Thin, and et al. 2020. "Portable Rabies Virus Sequencing in Canine Rabies Endemic Countries Using the Oxford Nanopore MinION" Viruses 12, no. 11: 1255. https://doi.org/10.3390/v12111255
APA StyleGigante, C. M., Yale, G., Condori, R. E., Costa, N. C., Long, N. V., Minh, P. Q., Chuong, V. D., Tho, N. D., Thanh, N. T., Thin, N. X., Hanh, N. T. H., Wambura, G., Ade, F., Mito, O., Chuchu, V., Muturi, M., Mwatondo, A., Hampson, K., Thumbi, S. M., ... Li, Y. (2020). Portable Rabies Virus Sequencing in Canine Rabies Endemic Countries Using the Oxford Nanopore MinION. Viruses, 12(11), 1255. https://doi.org/10.3390/v12111255