System-Based Approaches to Delineate the Antiviral Innate Immune Landscape


{kind=link}
{kind=link}
Abstract
1. Introduction
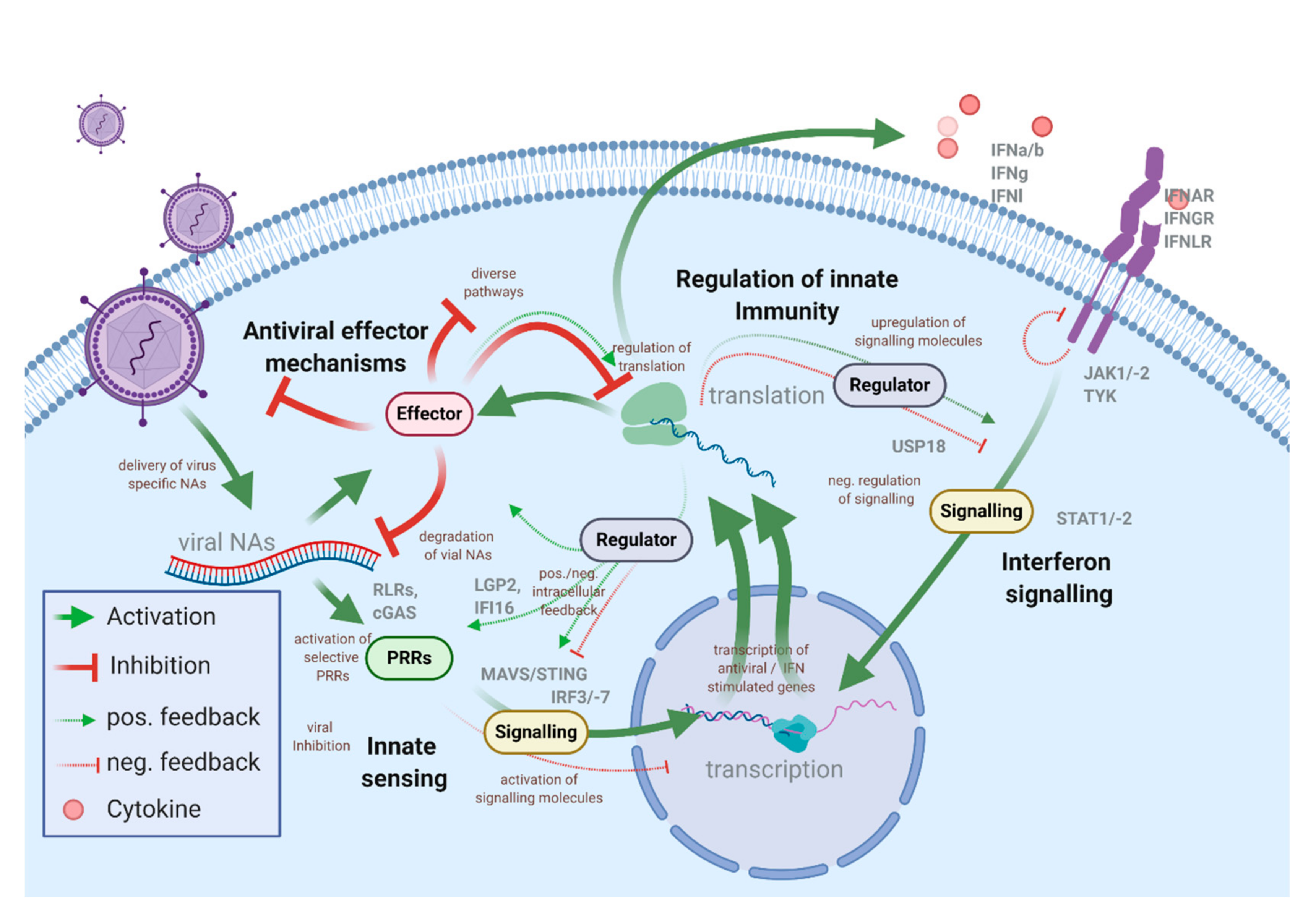
1.1. The Intracellular Antiviral Responses
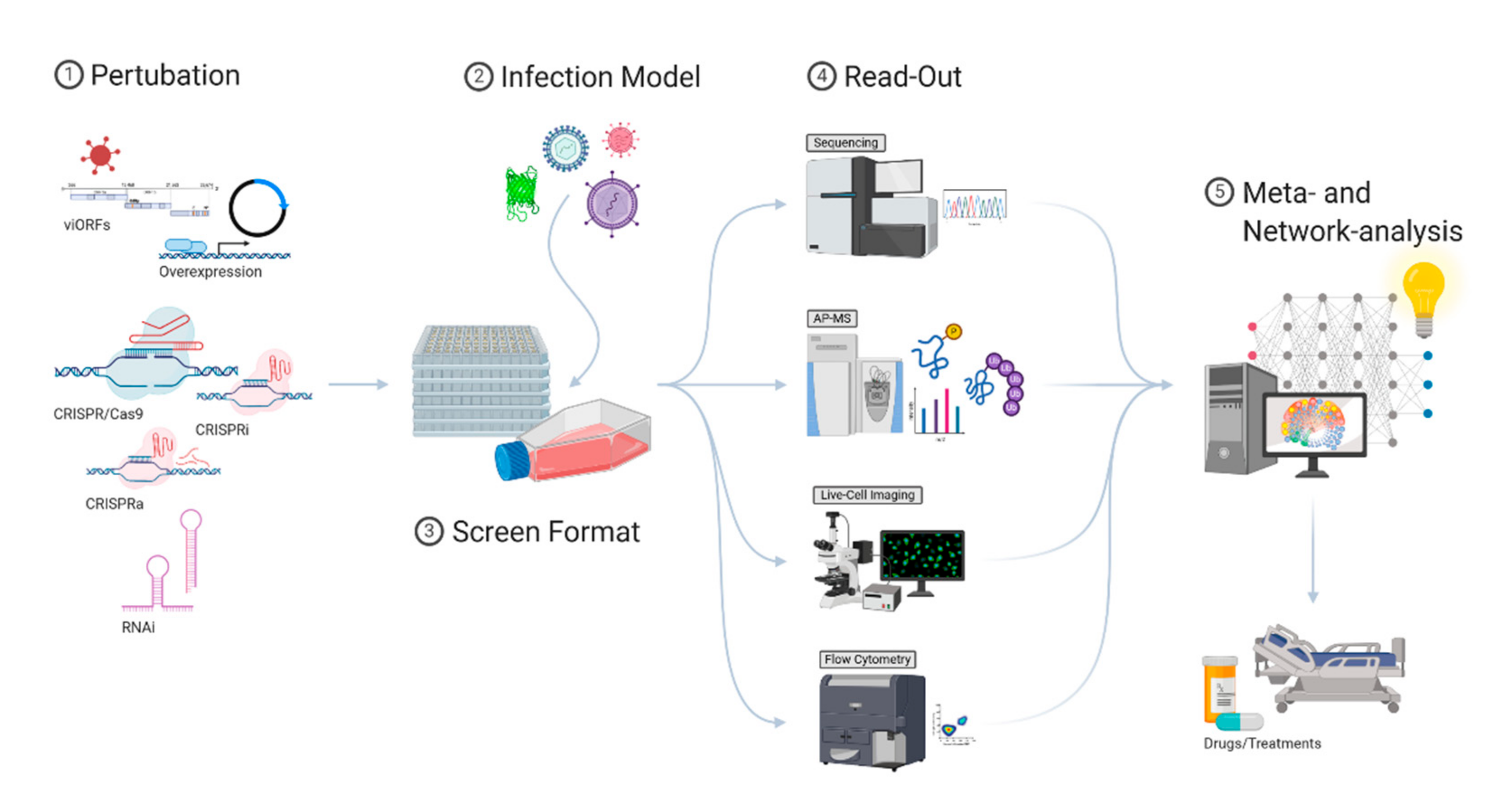
1.2. Experimental Systems to Study ISG Responses
2. Differential Gene Expression Analysis
3. Genetic Screens
3.1. Gain-of-Function Screens
3.2. Loss-of-Function Screens
3.2.1. RNA Interference
3.2.2. Haploid Screens
3.2.3. CRISPR/Cas9
3.3. Validation of Primary Screens
4. Assay Design and Screening Format
4.1. Choosing a Target Gene Library
4.2. The State of the Cell: Requirement of Pre-Stimulation to Allow Appropriate Experimental Conditions
4.3. Pooled Screens
4.4. Arrayed Screens
5. Interaction Screens and Mass-Spectrometry-Based Screens
5.1. Interaction Screens
5.2. Proteomic Approaches
5.3. Virus as Baits
5.4. Secretomics
6. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A. Box 1 (Methodology)
- RNA Interference (RNAi).
- Haploid Genetic Screens.
- CRISPR/Cas9.
- CRISPRa/i.
References
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Sanin, D.E.; Everts, B.; Chen, Q.; Qiu, J.; Buck, M.D.; Patterson, A.; Smith, A.M.; Chang, C.-H.; Liu, Z.; et al. Type 1 Interferons Induce Changes in Core Metabolism that Are Critical for Immune Function. Immunity 2016, 44, 1325–1336. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Kawai, T.; Akira, S. Pathogen recognition by the innate immune system. Int. Rev. Immunol. 2011, 30, 16–34. [Google Scholar] [CrossRef] [PubMed]
- Schlee, M.; Hartmann, G. Discriminating self from non-self in nucleic acid sensing. Nat. Rev. Immunol. 2016, 16, 566–580. [Google Scholar] [CrossRef] [PubMed]
- Arimoto, K.-I.; Miyauchi, S.; Stoner, S.A.; Fan, J.-B.; Zhang, D.-E. Negative regulation of type I IFN signaling. J. Leukoc. Biol. 2018, 103. [Google Scholar] [CrossRef] [PubMed]
- Blach-Olszewska, Z.; Leszek, J. Mechanisms of over-activated innate immune system regulation in autoimmune and neurodegenerative disorders. Neuropsychiatr. Dis. Treat. 2007, 3, 365–372. [Google Scholar]
- Liu, J.; Cao, X. Cellular and molecular regulation of innate inflammatory responses. Cell. Mol. Immunol. 2016, 13, 711–721. [Google Scholar] [CrossRef]
- Borden, E.C.; Sen, G.C.; Uze, G.; Silverman, R.H.; Ransohoff, R.M.; Foster, G.R.; Stark, G.R. Interferons at age 50: Past, current and future impact on biomedicine. Nat. Rev. Drug Discov. 2007, 6, 975–990. [Google Scholar] [CrossRef]
- Der, S.D.; Zhou, A.; Williams, B.R.; Silverman, R.H. Identification of genes differentially regulated by interferon alpha, beta, or gamma using oligonucleotide arrays. Proc. Natl. Acad. Sci. USA 1998, 95, 15623–15628. [Google Scholar] [CrossRef]
- Zhang, X.; Yang, W.; Wang, X.; Zhang, X.; Tian, H.; Deng, H.; Zhang, L.; Gao, G. Identification of new type I interferon-stimulated genes and investigation of their involvement in IFN-β activation. Protein Cell 2018, 9, 799–807. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef] [PubMed]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20, 1445. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Xu, L.; Su, J.; Peppelenbosch, M.P.; Pan, Q. Transcriptional Regulation of Antiviral Interferon-Stimulated Genes. Trends Microbiol. 2017, 25, 573–584. [Google Scholar] [CrossRef] [PubMed]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed]
- Nishitsuji, H.; Ujino, S.; Yoshio, S.; Sugiyama, M.; Mizokami, M.; Kanto, T.; Shimotohno, K. Long noncoding RNA #32 contributes to antiviral responses by controlling interferon-stimulated gene expression. Proc. Natl. Acad. Sci. USA 2016, 113, 10388–10393. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.E.; Hughes, J.; Gu, Q.; Behdenna, A.; Singer, J.B.; Dennis, T.; Orton, R.J.; Varela, M.; Gifford, R.J.; Wilson, S.J.; et al. Fundamental properties of the mammalian innate immune system revealed by multispecies comparison of type I interferon responses. PLoS Biol. 2017, 15, e2004086. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, G.; Kuzmanovic, T.; Zhang, Y.; Peter, C.B.; Veleeparambil, M.; Chakravarti, R.; Sen, G.C.; Chattopadhyay, S. A new mechanism of interferon’s antiviral action: Induction of autophagy, essential for paramyxovirus replication, is inhibited by the interferon stimulated gene, TDRD7. PLoS Pathog. 2018, 14, e1006877. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Zang, T.; Bieniasz, P.D. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 2008, 451, 425–430. [Google Scholar] [CrossRef]
- Neil, S.J.D.; Sandrin, V.; Sundquist, W.I.; Bieniasz, P.D. An Interferon-α-Induced Tethering Mechanism Inhibits HIV-1 and Ebola Virus Particle Release but Is Counteracted by the HIV-1 Vpu Protein. Cell Host Microbe 2007, 2, 193–203. [Google Scholar] [CrossRef]
- Yao, H.; Dittmann, M.; Peisley, A.; Hoffmann, H.-H.; Gilmore, R.H.; Schmidt, T.; Schmidt-Burgk, J.; Hornung, V.; Rice, C.M.; Hur, S. ATP-dependent effector-like functions of RIG-I-like receptors. Mol. Cell 2015, 58, 541–548. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed]
- Bruns, A.M.; Leser, G.P.; Lamb, R.A.; Horvath, C.M. The innate immune sensor LGP2 activates antiviral signaling by regulating MDA5-RNA interaction and filament assembly. Mol. Cell 2014, 55, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef] [PubMed]
- Jønsson, K.L.; Laustsen, A.; Krapp, C.; Skipper, K.A.; Thavachelvam, K.; Hotter, D.; Egedal, J.H.; Kjolby, M.; Mohammadi, P.; Prabakaran, T.; et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat. Commun. 2017, 8, 14391. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Liau, N.P.D.; Laktyushin, A.; Lucet, I.S.; Murphy, J.M.; Yao, S.; Whitlock, E.; Callaghan, K.; Nicola, N.A.; Kershaw, N.J.; Babon, J.J. The molecular basis of JAK/STAT inhibition by SOCS1. Nat. Commun. 2018, 9, 1558. [Google Scholar] [CrossRef]
- Blumer, T.; Coto-Llerena, M.; Duong, F.H.T.; Heim, M.H. SOCS1 is an inducible negative regulator of interferon λ (IFN-λ)–induced gene expression in vivo. J. Biol. Chem. 2017, 292, 17928–17938. [Google Scholar] [CrossRef]
- Akhtar, L.N.; Benveniste, E.N. Viral exploitation of host SOCS protein functions. J. Virol. 2011, 85, 1912–1921. [Google Scholar] [CrossRef]
- Liu, S.; Cai, X.; Wu, J.; Cong, Q.; Chen, X.; Li, T.; Du, F.; Ren, J.; Wu, Y.-T.; Grishin, N.V.; et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 2015, 347, aaa2630. [Google Scholar] [CrossRef]
- Ablasser, A.; Hur, S. Regulation of cGAS- and RLR-mediated immunity to nucleic acids. Nature immunology 2020, 21, 17–29. [Google Scholar] [CrossRef]
- Willemsen, J.; Wicht, O.; Wolanski, J.C.; Baur, N.; Bastian, S.; Haas, D.A.; Matula, P.; Knapp, B.; Meyniel-Schicklin, L.; Wang, C.; et al. Phosphorylation-Dependent Feedback Inhibition of RIG-I by DAPK1 Identified by Kinome-wide siRNA Screening. Mol. Cell 2017, 65, 403–415. [Google Scholar] [CrossRef] [PubMed]
- Pervolaraki, K.; Rastgou-Talemi, S.; Albrecht, D.; Bormann, F.; Bamford, C.; Mendoza, J.L.; Garcia, K.C.; McLauchlan, J.; Höfer, T.; Stanifer, M.L.; et al. Differential induction of interferon stimulated genes between type I and type III interferons is independent of interferon receptor abundance. PLoS Pathog. 2018, 14, e1007420. [Google Scholar] [CrossRef]
- Voigt, E.A.; Yin, J. Kinetic Differences and Synergistic Antiviral Effects Between Type I and Type III Interferon Signaling Indicate Pathway Independence. J. Interferon Cytokine Res. 2015, 35, 734–747. [Google Scholar] [CrossRef] [PubMed]
- Pommerenke, C.; Wilk, E.; Srivastava, B.; Schulze, A.; Novoselova, N.; Geffers, R.; Schughart, K. Global transcriptome analysis in influenza-infected mouse lungs reveals the kinetics of innate and adaptive host immune responses. PLoS ONE 2012, 7, e41169. [Google Scholar] [CrossRef] [PubMed]
- Michalska, A.; Blaszczyk, K.; Wesoly, J.; Bluyssen, H.A.R. A Positive Feedback Amplifier Circuit That Regulates Interferon (IFN)-Stimulated Gene Expression and Controls Type I and Type II IFN Responses. Front. Immunol. 2018, 9, 1135. [Google Scholar] [CrossRef]
- Larner, A.C.; Chaudhuri, A.; Darnell, J.E. Transcriptional induction by interferon. New protein(s) determine the extent and length of the induction. J. Biol. Chem. 1986, 261, 453–459. [Google Scholar]
- Lumb, J.H.; Li, Q.; Popov, L.M.; Ding, S.; Keith, M.T.; Merrill, B.D.; Greenberg, H.B.; Li, J.B.; Carette, J.E. DDX6 Represses Aberrant Activation of Interferon-Stimulated Genes. Cell Rep. 2017, 20, 819–831. [Google Scholar] [CrossRef]
- Thompson, S.L.; Compton, D.A. Chromosomes and cancer cells. Chromosome Res. 2011, 19, 433–444. [Google Scholar] [CrossRef]
- McCormick, D.; Lin, Y.-T.; Grey, F. Identification of Host Factors Involved in Human Cytomegalovirus Replication, Assembly, and Egress Using a Two-Step Small Interfering RNA Screen. mBio 2018, 9. [Google Scholar] [CrossRef]
- Konno, H.; Yamauchi, S.; Berglund, A.; Putney, R.M.; Mulé, J.J.; Barber, G.N. Suppression of STING signaling through epigenetic silencing and missense mutation impedes DNA damage mediated cytokine production. Oncogene 2018, 37, 2037–2051. [Google Scholar] [CrossRef]
- Sun, B.; Sundström, K.B.; Chew, J.J.; Bist, P.; Gan, E.S.; Tan, H.C.; Goh, K.C.; Chawla, T.; Tang, C.K.; Ooi, E.E. Dengue virus activates cGAS through the release of mitochondrial DNA. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, M.; Koshiba, T.; Ichinohe, T. Influenza A virus M2 protein triggers mitochondrial DNA-mediated antiviral immune responses. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Khoury-Hanold, W.; Staron, M.; Tal, M.C.; Pineda, C.M.; Lang, S.M.; Bestwick, M.; Duguay, B.A.; Raimundo, N.; MacDuff, D.A.; et al. Mitochondrial DNA stress primes the antiviral innate immune response. Nature 2015, 520, 553–557. [Google Scholar] [CrossRef]
- Rivas, C.; Aaronson, S.A.; Munoz-Fontela, C. Dual Role of p53 in Innate Antiviral Immunity. Viruses 2010, 2, 298–313. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Fontela, C.; Pazos, M.; Delgado, I.; Murk, W.; Mungamuri, S.K.; Lee, S.W.; García-Sastre, A.; Moran, T.M.; Aaronson, S.A. p53 serves as a host antiviral factor that enhances innate and adaptive immune responses to influenza A virus. J. Immunol. 2011, 187, 6428–6436. [Google Scholar] [CrossRef]
- Enache, O.M.; Rendo, V.; Abdusamad, M.; Lam, D.; Davison, D.; Pal, S.; Currimjee, N.; Hess, J.; Pantel, S.; Nag, A.; et al. Cas9 activates the p53 pathway and selects for p53-inactivating mutations. Nat. Genet. 2020, 52, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Thi-Nhu-Thao, T.; Labroussaa, F.; Ebert, N.; V’kovski, P.; Stalder, H.; Portmann, J.; Kelly, J.; Steiner, S.; Holwerda, M.; Kratzel, A.; et al. Rapid reconstruction of SARS-CoV-2 using a synthetic genomics platform. Nature 2020, 582, 561–565. [Google Scholar] [CrossRef]
- Kuroda, M.; Halfmann, P.J.; Hill-Batorski, L.; Ozawa, M.; Lopes, T.J.S.; Neumann, G.; Schoggins, J.W.; Rice, C.M.; Kawaoka, Y. Identification of interferon-stimulated genes that attenuate Ebola virus infection. Nat. Commun. 2020, 11, 2953. [Google Scholar] [CrossRef]
- Halfmann, P.; Kim, J.H.; Ebihara, H.; Noda, T.; Neumann, G.; Feldmann, H.; Kawaoka, Y. Generation of biologically contained Ebola viruses. Proc. Natl. Acad. Sci. USA 2008, 105, 1129–1133. [Google Scholar] [CrossRef]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Moll, H.P.; Maier, T.; Zommer, A.; Lavoie, T.; Brostjan, C. The differential activity of interferon-α subtypes is consistent among distinct target genes and cell types. Cytokine 2011, 53, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Moraga, I.; Levin, D.; Krutzik, P.O.; Podoplelova, Y.; Trejo, A.; Lee, C.; Yarden, G.; Vleck, S.E.; Glenn, J.S.; et al. Structural linkage between ligand discrimination and receptor activation by type I interferons. Cell 2011, 146, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Bolen, C.R.; Ding, S.; Robek, M.D.; Kleinstein, S.H. Dynamic expression profiling of type I and type III interferon-stimulated hepatocytes reveals a stable hierarchy of gene expression. Hepatology 2014, 59, 1262–1272. [Google Scholar] [CrossRef]
- Steuerman, Y.; Cohen, M.; Peshes-Yaloz, N.; Valadarsky, L.; Cohn, O.; David, E.; Frishberg, A.; Mayo, L.; Bacharach, E.; Amit, I.; et al. Dissection of Influenza Infection In Vivo by Single-Cell RNA Sequencing. Cell Syst. 2018, 6, 679–691. [Google Scholar] [CrossRef] [PubMed]
- Zanini, F.; Robinson, M.L.; Croote, D.; Sahoo, M.K.; Sanz, A.M.; Ortiz-Lasso, E.; Albornoz, L.L.; Rosso, F.; Montoya, J.G.; Goo, L.; et al. Virus-inclusive single-cell RNA sequencing reveals the molecular signature of progression to severe dengue. Proc. Natl. Acad. Sci. USA 2018, 115, E12363–E12369. [Google Scholar] [CrossRef]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; Wilson, S.J.; Panis, M.; Murphy, M.Y.; Jones, C.T.; Bieniasz, P.; Rice, C.M. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 2011, 472, 481–485. [Google Scholar] [CrossRef] [PubMed]
- Schoggins, J.W.; MacDuff, D.A.; Imanaka, N.; Gainey, M.D.; Shrestha, B.; Eitson, J.L.; Mar, K.B.; Richardson, R.B.; Ratushny, A.V.; Litvak, V.; et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature 2014, 505, 691–695. [Google Scholar] [CrossRef]
- Riley, J.S.; Tait, S.W. Mitochondrial DNA in inflammation and immunity. EMBO Rep. 2020, 21, e49799. [Google Scholar] [CrossRef]
- Seth, R.B.; Sun, L.; Ea, C.-K.; Chen, Z.J. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell 2005, 122, 669–682. [Google Scholar] [CrossRef]
- Biacchesi, S.; Mérour, E.; Lamoureux, A.; Bernard, J.; Brémont, M. Both STING and MAVS fish orthologs contribute to the induction of interferon mediated by RIG-I. PLoS ONE 2012, 7, e47737. [Google Scholar] [CrossRef]
- Stremlau, M.; Owens, C.M.; Perron, M.J.; Kiessling, M.; Autissier, P.; Sodroski, J. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 2004, 427, 848–853. [Google Scholar] [CrossRef] [PubMed]
- Kane, M.; Zang, T.M.; Rihn, S.J.; Zhang, F.; Kueck, T.; Alim, M.; Schoggins, J.; Rice, C.M.; Wilson, S.J.; Bieniasz, P.D. Identification of Interferon-Stimulated Genes with Antiretroviral Activity. Cell Host Microbe 2016, 20, 392–405. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Wickenhagen, A.; Turnbull, M.L.; Rezelj, V.V.; Kreher, F.; Tilston-Lunel, N.L.; Slack, G.S.; Brennan, B.; Koudriakova, E.; Shaw, A.E.; et al. Interferon-Stimulated Gene (ISG)-Expression Screening Reveals the Specific Antibunyaviral Activity of ISG20. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Lassnig, C.; Eberle, C.-A.; Górna, M.W.; Baumann, C.L.; Burkard, T.R.; Bürckstümmer, T.; Stefanovic, A.; Krieger, S.; Bennett, K.L.; et al. IFIT1 is an antiviral protein that recognizes 5′-triphosphate RNA. Nat. Immunol. 2011, 12, 624–630. [Google Scholar] [CrossRef]
- Johnson, B.; VanBlargan, L.A.; Xu, W.; White, J.P.; Shan, C.; Shi, P.-Y.; Zhang, R.; Adhikari, J.; Gross, M.L.; Leung, D.W.; et al. Human IFIT3 Modulates IFIT1 RNA Binding Specificity and Protein Stability. Immunity 2018, 48, 487–499. [Google Scholar] [CrossRef]
- Fellmann, C.; Lowe, S.W. Stable RNA interference rules for silencing. Nat. Cell Biol. 2014, 16, 10–18. [Google Scholar] [CrossRef]
- Schuster, A.; Erasimus, H.; Fritah, S.; Nazarov, P.V.; van Dyck, E.; Niclou, S.P.; Golebiewska, A. RNAi/CRISPR Screens: From a Pool to a Valid Hit. Trends Biotechnol. 2019, 37, 38–55. [Google Scholar] [CrossRef]
- Boettcher, M.; McManus, M.T. Choosing the Right Tool for the Job: RNAi, TALEN, or CRISPR. Mol. Cell 2015, 58, 575–585. [Google Scholar] [CrossRef]
- Carthew, R.W.; Sontheimer, E.J. Origins and Mechanisms of miRNAs and siRNAs. Cell 2009, 136, 642–655. [Google Scholar] [CrossRef]
- Agarwal, V.; Bell, G.W.; Nam, J.-W.; Bartel, D.P. Predicting effective microRNA target sites in mammalian mRNAs. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Riba, A.; Emmenlauer, M.; Chen, A.; Sigoillot, F.; Cong, F.; Dehio, C.; Jenkins, J.; Zavolan, M. Explicit Modeling of siRNA-Dependent On- and Off-Target Repression Improves the Interpretation of Screening Results. Cell Syst. 2017, 4, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Pillay, S.; Meyer, N.L.; Puschnik, A.S.; Davulcu, O.; Diep, J.; Ishikawa, Y.; Jae, L.T.; Wosen, J.E.; Nagamine, C.M.; Chapman, M.S.; et al. An essential receptor for adeno-associated virus infection. Nature 2016, 530, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Raaben, M.; Wong, A.C.; Herbert, A.S.; Obernosterer, G.; Mulherkar, N.; Kuehne, A.I.; Kranzusch, P.J.; Griffin, A.M.; Ruthel, G.; et al. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. [Google Scholar] [CrossRef]
- Jae, L.T.; Raaben, M.; Herbert, A.S.; Kuehne, A.I.; Wirchnianski, A.S.; Soh, T.K.; Stubbs, S.H.; Janssen, H.; Damme, M.; Saftig, P.; et al. Virus entry. Lassa virus entry requires a trigger-induced receptor switch. Science 2014, 344, 1506–1510. [Google Scholar] [CrossRef] [PubMed]
- Staring, J.; von Castelmur, E.; Blomen, V.A.; van den Hengel, L.G.; Brockmann, M.; Baggen, J.; Thibaut, H.J.; Nieuwenhuis, J.; Janssen, H.; van Kuppeveld, F.J.M.; et al. PLA2G16 represents a switch between entry and clearance of Picornaviridae. Nature 2017, 541, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Carette, J.E.; Guimaraes, C.P.; Varadarajan, M.; Park, A.S.; Wuethrich, I.; Godarova, A.; Kotecki, M.; Cochran, B.H.; Spooner, E.; Ploegh, H.L.; et al. Haploid genetic screens in human cells identify host factors used by pathogens. Science 2009, 326, 1231–1235. [Google Scholar] [CrossRef]
- Riblett, A.M.; Blomen, V.A.; Jae, L.T.; Altamura, L.A.; Doms, R.W.; Brummelkamp, T.R.; Wojcechowskyj, J.A. A Haploid Genetic Screen Identifies Heparan Sulfate Proteoglycans Supporting Rift Valley Fever Virus Infection. J. Virol. 2016, 90, 1414–1423. [Google Scholar] [CrossRef]
- Luteijn, R.D.; van Diemen, F.; Blomen, V.A.; Boer, I.G.J.; Manikam-Sadasivam, S.; van Kuppevelt, T.H.; Drexler, I.; Brummelkamp, T.R.; Lebbink, R.J.; Wiertz, E.J. A Genome-Wide Haploid Genetic Screen Identifies Heparan Sulfate-Associated Genes and the Macropinocytosis Modulator TMED10 as Factors Supporting Vaccinia Virus Infection. J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Doench, J.G.; Fusi, N.; Sullender, M.; Hegde, M.; Vaimberg, E.W.; Donovan, K.F.; Smith, I.; Tothova, Z.; Wilen, C.; Orchard, R.; et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016, 34, 184–191. [Google Scholar] [CrossRef]
- Munoz, D.M.; Cassiani, P.J.; Li, L.; Billy, E.; Korn, J.M.; Jones, M.D.; Golji, J.; Ruddy, D.A.; Yu, K.; McAllister, G.; et al. CRISPR Screens Provide a Comprehensive Assessment of Cancer Vulnerabilities but Generate False-Positive Hits for Highly Amplified Genomic Regions. Cancer Discov. 2016, 6, 900–913. [Google Scholar] [CrossRef] [PubMed]
- Duan, J.; Lu, G.; Xie, Z.; Lou, M.; Luo, J.; Guo, L.; Zhang, Y. Genome-wide identification of CRISPR/Cas9 off-targets in human genome. Cell Res. 2014, 24, 1009–1012. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676. [Google Scholar] [CrossRef] [PubMed]
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014, 32, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.Q.; Zheng, Z.; Nguyen, N.T.; Liebers, M.; Topkar, V.V.; Thapar, V.; Wyvekens, N.; Khayter, C.; Iafrate, A.J.; Le, L.P.; et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015, 33, 187–197. [Google Scholar] [CrossRef]
- Joung, J.; Konermann, S.; Gootenberg, J.S.; Abudayyeh, O.O.; Platt, R.J.; Brigham, M.D.; Sanjana, N.E.; Zhang, F. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat. Protoc. 2017, 12, 828–863. [Google Scholar] [CrossRef]
- Lee, C.-H.; Griffiths, S.; Digard, P.; Pham, N.; Auer, M.; Haas, J.; Grey, F. Asparagine Deprivation Causes a Reversible Inhibition of Human Cytomegalovirus Acute Virus Replication. mBio 2019, 10. [Google Scholar] [CrossRef]
- van Asten, S.D.; Raaben, M.; Nota, B.; Spaapen, R.M. Secretome Screening Reveals Fibroblast Growth Factors as Novel Inhibitors of Viral Replication. J. Virol. 2018, 92. [Google Scholar] [CrossRef]
- Puschnik, A.S.; Majzoub, K.; Ooi, Y.S.; Carette, J.E. A CRISPR toolbox to study virus-host interactions. Nat. Rev. Microbiol. 2017, 15, 351–364. [Google Scholar] [CrossRef]
- Chou, Y.-C.; Lai, M.M.; Wu, Y.-C.; Hsu, N.-C.; Jeng, K.-S.; Su, W.-C. Variations in genome-wide RNAi screens: Lessons from influenza research. J. Clin. Bioinforma. 2015, 5, 2. [Google Scholar] [CrossRef]
- Hao, L.; He, Q.; Wang, Z.; Craven, M.; Newton, M.A.; Ahlquist, P. Limited agreement of independent RNAi screens for virus-required host genes owes more to false-negative than false-positive factors. PLoS Comput. Biol. 2013, 9, e1003235. [Google Scholar] [CrossRef] [PubMed]
- Sessions, O.M.; Barrows, N.J.; Souza-Neto, J.A.; Robinson, T.J.; Hershey, C.L.; Rodgers, M.A.; Ramirez, J.L.; Dimopoulos, G.; Yang, P.L.; Pearson, J.L.; et al. Discovery of insect and human dengue virus host factors. Nature 2009, 458, 1047–1050. [Google Scholar] [CrossRef]
- Savidis, G.; McDougall, W.M.; Meraner, P.; Perreira, J.M.; Portmann, J.M.; Trincucci, G.; John, S.P.; Aker, A.M.; Renzette, N.; Robbins, D.R.; et al. Identification of Zika Virus and Dengue Virus Dependency Factors using Functional Genomics. Cell Rep. 2016, 16, 232–246. [Google Scholar] [CrossRef]
- Filone, C.M.; Dower, K.; Cowley, G.S.; Hensley, L.E.; Connor, J.H. Probing the virus host interaction in high containment: An approach using pooled short hairpin RNA. Assay Drug Dev. Technol. 2015, 13, 34–43. [Google Scholar] [CrossRef]
- Martin, S.; Chiramel, A.I.; Schmidt, M.L.; Chen, Y.-C.; Whitt, N.; Watt, A.; Dunham, E.C.; Shifflett, K.; Traeger, S.; Leske, A.; et al. A genome-wide siRNA screen identifies a druggable host pathway essential for the Ebola virus life cycle. Genome Med. 2018, 10, 58. [Google Scholar] [CrossRef] [PubMed]
- Tai, A.W.; Benita, Y.; Peng, L.F.; Kim, S.-S.; Sakamoto, N.; Xavier, R.J.; Chung, R.T. A functional genomic screen identifies cellular cofactors of hepatitis C virus replication. Cell Host Microbe 2009, 5, 298–307. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Brass, A.L.; Ng, A.; Hu, Z.; Xavier, R.J.; Liang, T.J.; Elledge, S.J. A genome-wide genetic screen for host factors required for hepatitis C virus propagation. Proc. Natl. Acad. Sci. USA 2009, 106, 16410–16415. [Google Scholar] [CrossRef] [PubMed]
- Ali, H.; Mano, M.; Braga, L.; Naseem, A.; Marini, B.; Vu, D.M.; Collesi, C.; Meroni, G.; Lusic, M.; Giacca, M. Cellular TRIM33 restrains HIV-1 infection by targeting viral integrase for proteasomal degradation. Nat. Commun. 2019, 10, 926. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef]
- König, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.C.; Irelan, J.T.; Chiang, C.-Y.; Tu, B.P.; de Jesus, P.D.; Lilley, C.E.; et al. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.-M.; Zhang, X.-Y.; Zhang, Y.-Z.; Liu, L.; Lu, H.-Z. A high throughput RNAi screen reveals determinants of HIV-1 activity in host kinases. Int. J. Clin. Exp. Pathol. 2014, 7, 2229–2237. [Google Scholar] [PubMed]
- König, R.; Stertz, S.; Zhou, Y.; Inoue, A.; Hoffmann, H.-H.; Bhattacharyya, S.; Alamares, J.G.; Tscherne, D.M.; Ortigoza, M.B.; Liang, Y.; et al. Human host factors required for influenza virus replication. Nature 2010, 463, 813–817. [Google Scholar] [CrossRef] [PubMed]
- Su, W.-C.; Chen, Y.-C.; Tseng, C.-H.; Hsu, P.W.-C.; Tung, K.-F.; Jeng, K.-S.; Lai, M.M.C. Pooled RNAi screen identifies ubiquitin ligase Itch as crucial for influenza A virus release from the endosome during virus entry. Proc. Natl. Acad. Sci. USA 2013, 110, 17516–17521. [Google Scholar] [CrossRef]
- Hao, L.; Sakurai, A.; Watanabe, T.; Sorensen, E.; Nidom, C.A.; Newton, M.A.; Ahlquist, P.; Kawaoka, Y. Drosophila RNAi screen identifies host genes important for influenza virus replication. Nature 2008, 454, 890–893. [Google Scholar] [CrossRef]
- Karlas, A.; Machuy, N.; Shin, Y.; Pleissner, K.-P.; Artarini, A.; Heuer, D.; Becker, D.; Khalil, H.; Ogilvie, L.A.; Hess, S.; et al. Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010, 463, 818–822. [Google Scholar] [CrossRef]
- Cheng, H.; Koning, K.; O’Hearn, A.; Wang, M.; Rumschlag-Booms, E.; Varhegyi, E.; Rong, L. A parallel genome-wide RNAi screening strategy to identify host proteins important for entry of Marburg virus and H5N1 influenza virus. Virol. J. 2015, 12, 194. [Google Scholar] [CrossRef]
- Panda, D.; Rose, P.P.; Hanna, S.L.; Gold, B.; Hopkins, K.C.; Lyde, R.B.; Marks, M.S.; Cherry, S. Genome-wide RNAi screen identifies SEC61A and VCP as conserved regulators of Sindbis virus entry. Cell Rep. 2013, 5, 1737–1748. [Google Scholar] [CrossRef]
- Rose, P.P.; Hanna, S.L.; Spiridigliozzi, A.; Wannissorn, N.; Beiting, D.P.; Ross, S.R.; Hardy, R.W.; Bambina, S.A.; Heise, M.T.; Cherry, S. Natural resistance-associated macrophage protein is a cellular receptor for sindbis virus in both insect and mammalian hosts. Cell Host Microbe 2011, 10, 97–104. [Google Scholar] [CrossRef]
- Krishnan, M.N.; Ng, A.; Sukumaran, B.; Gilfoy, F.D.; Uchil, P.D.; Sultana, H.; Brass, A.L.; Adametz, R.; Tsui, M.; Qian, F.; et al. RNA interference screen for human genes associated with West Nile virus infection. Nature 2008, 455, 242–245. [Google Scholar] [CrossRef]
- Ma, H.; Dang, Y.; Wu, Y.; Jia, G.; Anaya, E.; Zhang, J.; Abraham, S.; Choi, J.-G.; Shi, G.; Qi, L.; et al. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015, 12, 673–683. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Clohisey, S.M.; Chia, B.S.; Wang, B.; Cui, A.; Eisenhaure, T.; Schweitzer, L.D.; Hoover, P.; Parkinson, N.J.; Nachshon, A.; et al. Genome-wide CRISPR screen identifies host dependency factors for influenza A virus infection. Nat. Commun. 2020, 11, 164. [Google Scholar] [CrossRef] [PubMed]
- Han, J.; Perez, J.T.; Chen, C.; Li, Y.; Benitez, A.; Kandasamy, M.; Lee, Y.; Andrade, J.; tenOever, B.; Manicassamy, B. Genome-wide CRISPR/Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication. Cell Rep. 2018, 23, 596–607. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Muffat, J.; Omer-Javed, A.; Keys, H.R.; Lungjangwa, T.; Bosch, I.; Khan, M.; Virgilio, M.C.; Gehrke, L.; Sabatini, D.M.; et al. Genome-wide CRISPR screen for Zika virus resistance in human neural cells. Proc. Natl. Acad. Sci. USA 2019, 116, 9527–9532. [Google Scholar] [CrossRef]
- Wei, J.; Alfajaro, M.M.; Hanna, R.E.; DeWeirdt, P.C.; Strine, M.S.; Lu-Culligan, W.J.; Zhang, S.-M.; Graziano, V.R.; Schmitz, C.O.; Chen, J.S.; et al. Genome-wide CRISPR screen reveals host genes that regulate SARS-CoV-2 infection. bioRxiv 2020. [Google Scholar] [CrossRef]
- Thamamongood, T.; Aebischer, A.; Wagner, V.; Chang, M.W.; Elling, R.; Benner, C.; García-Sastre, A.; Kochs, G.; Beer, M.; Schwemmle, M. A Genome-Wide CRISPR-Cas9 Screen Reveals the Requirement of Host Cell Sulfation for Schmallenberg Virus Infection. J. Virol. 2020, 94. [Google Scholar] [CrossRef]
- Roesch, F.; OhAinle, M.; Emerman, M. A CRISPR screen for factors regulating SAMHD1 degradation identifies IFITMs as potent inhibitors of lentiviral particle delivery. Retrovirology 2018, 15, 26. [Google Scholar] [CrossRef]
- Richardson, R.B.; Ohlson, M.B.; Eitson, J.L.; Kumar, A.; McDougal, M.B.; Boys, I.N.; Mar, K.B.; de la Cruz-Rivera, P.C.; Douglas, C.; Konopka, G.; et al. A CRISPR screen identifies IFI6 as an ER-resident interferon effector that blocks flavivirus replication. Nat. Microbiol. 2018, 3, 1214–1223. [Google Scholar] [CrossRef]
- Varble, A.; Benitez, A.A.; Schmid, S.; Sachs, D.; Shim, J.V.; Rodriguez-Barrueco, R.; Panis, M.; Crumiller, M.; Silva, J.M.; Sachidanandam, R.; et al. An in vivo RNAi screening approach to identify host determinants of virus replication. Cell Host Microbe 2013, 14, 346–356. [Google Scholar] [CrossRef]
- Benaoudia, S.; Martin, A.; Puig-Gamez, M.; Gay, G.; Lagrange, B.; Cornut, M.; Krasnykov, K.; Claude, J.-B.; Bourgeois, C.F.; Hughes, S.; et al. A genome-wide screen identifies IRF2 as a key regulator of caspase-4 in human cells. EMBO Rep. 2019, 20, e48235. [Google Scholar] [CrossRef]
- Orvedahl, A.; McAllaster, M.R.; Sansone, A.; Dunlap, B.F.; Desai, C.; Wang, Y.-T.; Balce, D.R.; Luke, C.J.; Lee, S.; Orchard, R.C.; et al. Autophagy genes in myeloid cells counteract IFNγ-induced TNF-mediated cell death and fatal TNF-induced shock. Proc. Natl. Acad. Sci. USA 2019, 116, 16497–16506. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Diep, J.; Feng, N.; Ren, L.; Li, B.; Ooi, Y.S.; Wang, X.; Brulois, K.F.; Yasukawa, L.L.; Li, X.; et al. STAG2 deficiency induces interferon responses via cGAS-STING pathway and restricts virus infection. Nat. Commun. 2018, 9, 1485. [Google Scholar] [CrossRef]
- OhAinle, M.; Helms, L.; Vermeire, J.; Roesch, F.; Humes, D.; Basom, R.; Delrow, J.J.; Overbaugh, J.; Emerman, M. A virus-packageable CRISPR screen identifies host factors mediating interferon inhibition of HIV. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, K.; Bae, S.; Park, J.; Lee, C.-K.; Kim, M.; Kim, E.; Kim, M.; Kim, S.; Kim, C.; et al. CRISPR/Cas9-mediated gene knockout screens and target identification via whole-genome sequencing uncover host genes required for picornavirus infection. J. Biol. Chem. 2017, 292, 10664–10671. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Lu, T.; Emanuel, G.; Babcock, H.P.; Zhuang, X. Imaging-based pooled CRISPR screening reveals regulators of lncRNA localization. Proc. Natl. Acad. Sci. USA 2019, 116, 10842–10851. [Google Scholar] [CrossRef] [PubMed]
- Datlinger, P.; Rendeiro, A.F.; Schmidl, C.; Krausgruber, T.; Traxler, P.; Klughammer, J.; Schuster, L.C.; Kuchler, A.; Alpar, D.; Bock, C. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods 2017, 14, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Metzakopian, E.; Strong, A.; Iyer, V.; Hodgkins, A.; Tzelepis, K.; Antunes, L.; Friedrich, M.J.; Kang, Q.; Davidson, T.; Lamberth, J.; et al. Enhancing the genome editing toolbox: genome wide CRISPR arrayed libraries. Sci. Rep. 2017, 7, 2244. [Google Scholar] [CrossRef]
- Kim, H.S.; Lee, K.; Kim, S.-J.; Cho, S.; Shin, H.J.; Kim, C.; Kim, J.-S. Arrayed CRISPR screen with image-based assay reliably uncovers host genes required for coxsackievirus infection. Genome Res. 2018, 28, 859–868. [Google Scholar] [CrossRef]
- Fields, S.; Song, O. A novel genetic system to detect protein-protein interactions. Nature 1989, 340, 245–246. [Google Scholar] [CrossRef]
- McCraith, S.; Holtzman, T.; Moss, B.; Fields, S. Genome-wide analysis of vaccinia virus protein-protein interactions. Proc. Natl. Acad. Sci. USA 2000, 97, 4879–4884. [Google Scholar] [CrossRef]
- von Brunn, A.; Teepe, C.; Simpson, J.C.; Pepperkok, R.; Friedel, C.C.; Zimmer, R.; Roberts, R.; Baric, R.; Haas, J. Analysis of intraviral protein-protein interactions of the SARS coronavirus ORFeome. PLoS ONE 2007, 2, e459. [Google Scholar] [CrossRef]
- Flajolet, M.; Rotondo, G.; Daviet, L.; Bergametti, F.; Inchauspé, G.; Tiollais, P.; Transy, C.; Legrain, P. A genomic approach of the hepatitis C virus generates a protein interaction map. Gene 2000, 242, 369–379. [Google Scholar] [CrossRef]
- Fossum, E.; Friedel, C.C.; Rajagopala, S.V.; Titz, B.; Baiker, A.; Schmidt, T.; Kraus, T.; Stellberger, T.; Rutenberg, C.; Suthram, S.; et al. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 2009, 5, e1000570. [Google Scholar] [CrossRef] [PubMed]
- Uetz, P.; Dong, Y.-A.; Zeretzke, C.; Atzler, C.; Baiker, A.; Berger, B.; Rajagopala, S.V.; Roupelieva, M.; Rose, D.; Fossum, E.; et al. Herpesviral protein networks and their interaction with the human proteome. Science 2006, 311, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Calderwood, M.A.; Venkatesan, K.; Xing, L.; Chase, M.R.; Vazquez, A.; Holthaus, A.M.; Ewence, A.E.; Li, N.; Hirozane-Kishikawa, T.; Hill, D.E.; et al. Epstein-Barr virus and virus human protein interaction maps. Proc. Natl. Acad. Sci. USA 2007, 104, 7606–7611. [Google Scholar] [CrossRef] [PubMed]
- de Chassey, B.; Navratil, V.; Tafforeau, L.; Hiet, M.S.; Aublin-Gex, A.; Agaugué, S.; Meiffren, G.; Pradezynski, F.; Faria, B.F.; Chantier, T.; et al. Hepatitis C virus infection protein network. Mol. Syst. Biol. 2008, 4, 230. [Google Scholar] [CrossRef] [PubMed]
- Shapira, S.D.; Gat-Viks, I.; Shum, B.O.V.; Dricot, A.; de Grace, M.M.; Wu, L.; Gupta, P.B.; Hao, T.; Silver, S.J.; Root, D.E.; et al. A physical and regulatory map of host-influenza interactions reveals pathways in H1N1 infection. Cell 2009, 139, 1255–1267. [Google Scholar] [CrossRef]
- Chen, J.-Y.; Chen, W.-N.; Poon, K.-M.V.; Zheng, B.-J.; Lin, X.; Wang, Y.-X.; Wen, Y.-M. Interaction between SARS-CoV helicase and a multifunctional cellular protein (Ddx5) revealed by yeast and mammalian cell two-hybrid systems. Arch. Virol. 2009, 154, 507–512. [Google Scholar] [CrossRef]
- Zhang, L.; Villa, N.Y.; Rahman, M.M.; Smallwood, S.; Shattuck, D.; Neff, C.; Dufford, M.; Lanchbury, J.S.; Labaer, J.; McFadden, G. Analysis of vaccinia virus-host protein-protein interactions: Validations of yeast two-hybrid screenings. J. Proteome Res. 2009, 8, 4311–4318. [Google Scholar] [CrossRef]
- Moerdyk-Schauwecker, M.; Destephanis, D.; Hastie, E.; Grdzelishvili, V.Z. Detecting protein-protein interactions in vesicular stomatitis virus using a cytoplasmic yeast two hybrid system. J. Virol. Methods 2011, 173, 203–212. [Google Scholar] [CrossRef]
- Hubel, P.; Urban, C.; Bergant, V.; Schneider, W.M.; Knauer, B.; Stukalov, A.; Scaturro, P.; Mann, A.; Brunotte, L.; Hoffmann, H.H.; et al. A protein-interaction network of interferon-stimulated genes extends the innate immune system landscape. Nat. Immunol. 2019, 20, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Lum, K.K.; Cristea, I.M. Proteomic approaches to uncovering virus-host protein interactions during the progression of viral infection. Expert Rev. Proteom. 2016, 13, 325–340. [Google Scholar] [CrossRef]
- Greco, T.M.; Diner, B.A.; Cristea, I.M. The Impact of Mass Spectrometry-Based Proteomics on Fundamental Discoveries in Virology. Annu. Rev. Virol. 2014, 1, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Aebersold, R.; Mann, M. Mass-spectrometric exploration of proteome structure and function. Nature 2016, 537, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, M.; Hultquist, J.F.; Kaake, R.M.; Hüttenhain, R.; Krogan, N.J. A systems approach to infectious disease. Nat. Rev. Genet. 2020, 21, 339–354. [Google Scholar] [CrossRef]
- Andrejeva, J.; Childs, K.S.; Young, D.F.; Carlos, T.S.; Stock, N.; Goodbourn, S.; Randall, R.E. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc. Natl. Acad. Sci. USA 2004, 101, 17264–17269. [Google Scholar] [CrossRef] [PubMed]
- Pichlmair, A.; Kandasamy, K.; Alvisi, G.; Mulhern, O.; Sacco, R.; Habjan, M.; Binder, M.; Stefanovic, A.; Eberle, C.-A.; Goncalves, A.; et al. Viral immune modulators perturb the human molecular network by common and unique strategies. Nature 2012, 487, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Scaturro, P.; Stukalov, A.; Haas, D.A.; Cortese, M.; Draganova, K.; Płaszczyca, A.; Bartenschlager, R.; Götz, M.; Pichlmair, A. An orthogonal proteomic survey uncovers novel Zika virus host factors. Nature 2018, 561, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Stukalov, A.; Girault, V.; Grass, V.; Bergant, V.; Karayel, O.; Urban, C.; Haas, D.A.; Huang, Y.; Oubraham, L.; Wang, A.; et al. Multi-level proteomics reveals host-perturbation strategies of SARS-CoV-2 and SARS-CoV. bioRxiv 2020. [Google Scholar] [CrossRef]
- Jäger, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV-human protein complexes. Nature 2011, 481, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Koppenol-Raab, M.; Sjoelund, V.; Manes, N.P.; Gottschalk, R.A.; Dutta, B.; Benet, Z.L.; Fraser, I.D.C.; Nita-Lazar, A. Proteome and Secretome Analysis Reveals Differential Post-transcriptional Regulation of Toll-like Receptor Responses. Mol. Cell. Proteom. 2017, 16, S172–S186. [Google Scholar] [CrossRef] [PubMed]
- Meissner, F.; Scheltema, R.A.; Mollenkopf, H.-J.; Mann, M. Direct proteomic quantification of the secretome of activated immune cells. Science 2013, 340, 475–478. [Google Scholar] [CrossRef] [PubMed]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef] [PubMed]
- Wilson, R.C.; Doudna, J.A. Molecular mechanisms of RNA interference. Annu. Rev. Biophys. 2013, 42, 217–239. [Google Scholar] [CrossRef]
- Essletzbichler, P.; Konopka, T.; Santoro, F.; Chen, D.; Gapp, B.V.; Kralovics, R.; Brummelkamp, T.R.; Nijman, S.M.B.; Bürckstümmer, T. Megabase-scale deletion using CRISPR/Cas9 to generate a fully haploid human cell line. Genome Res. 2014, 24, 2059–2065. [Google Scholar] [CrossRef]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Brinkman, E.K.; Chen, T.; de Haas, M.; Holland, H.A.; Akhtar, W.; van Steensel, B. Kinetics and Fidelity of the Repair of Cas9-Induced Double-Strand DNA Breaks. Mol. Cell 2018, 70, 801–813. [Google Scholar] [CrossRef]
- Kurosaki, T.; Maquat, L.E. Nonsense-mediated mRNA decay in humans at a glance. J. Cell Sci. 2016, 129, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krey, K.; Babnis, A.W.; Pichlmair, A. System-Based Approaches to Delineate the Antiviral Innate Immune Landscape. Viruses 2020, 12, 1196. https://doi.org/10.3390/v12101196
Krey K, Babnis AW, Pichlmair A. System-Based Approaches to Delineate the Antiviral Innate Immune Landscape. Viruses. 2020; 12(10):1196. https://doi.org/10.3390/v12101196
Chicago/Turabian StyleKrey, Karsten, Aleksandra W. Babnis, and Andreas Pichlmair. 2020. "System-Based Approaches to Delineate the Antiviral Innate Immune Landscape" Viruses 12, no. 10: 1196. https://doi.org/10.3390/v12101196
APA StyleKrey, K., Babnis, A. W., & Pichlmair, A. (2020). System-Based Approaches to Delineate the Antiviral Innate Immune Landscape. Viruses, 12(10), 1196. https://doi.org/10.3390/v12101196