Genome Analysis of a Novel Clade b Betabaculovirus Isolated from the Legume Pest Matsumuraeses phaseoli (Lepidoptera: Tortricidae)



Abstract
1. Introduction
2. Materials and Methods
2.1. Virus Collection and Host Identification
2.2. Morphological Characterization of the Occlusion Bodies
2.3. Genomic DNA Sequencing and Sequence Analysis
2.4. Non-coding Region Analysis
2.5. Phylogenetic Analysis
3. Results and Discussion
3.1. The Host Determination and Virus Characterization
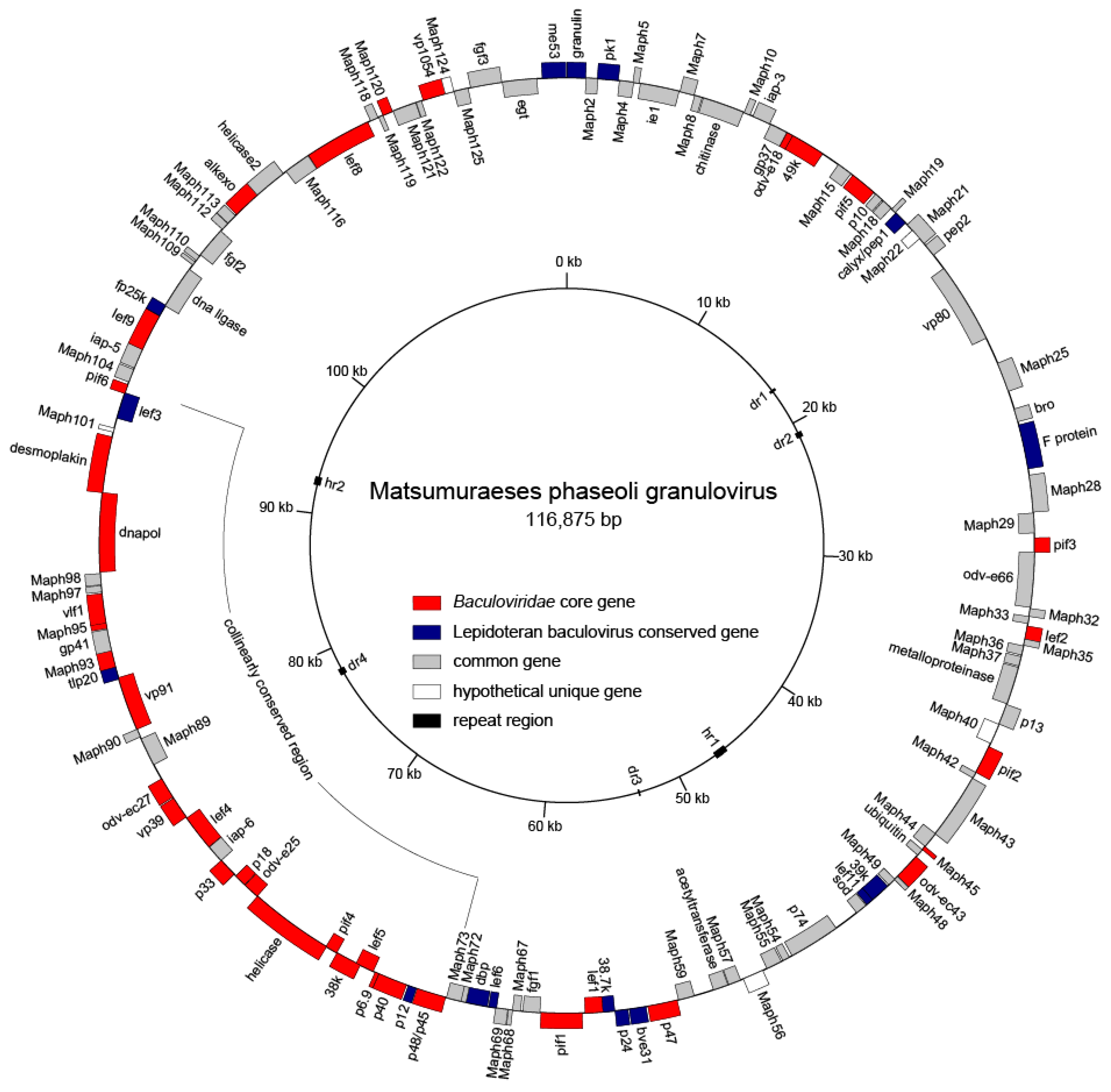
3.2. Genome Feature
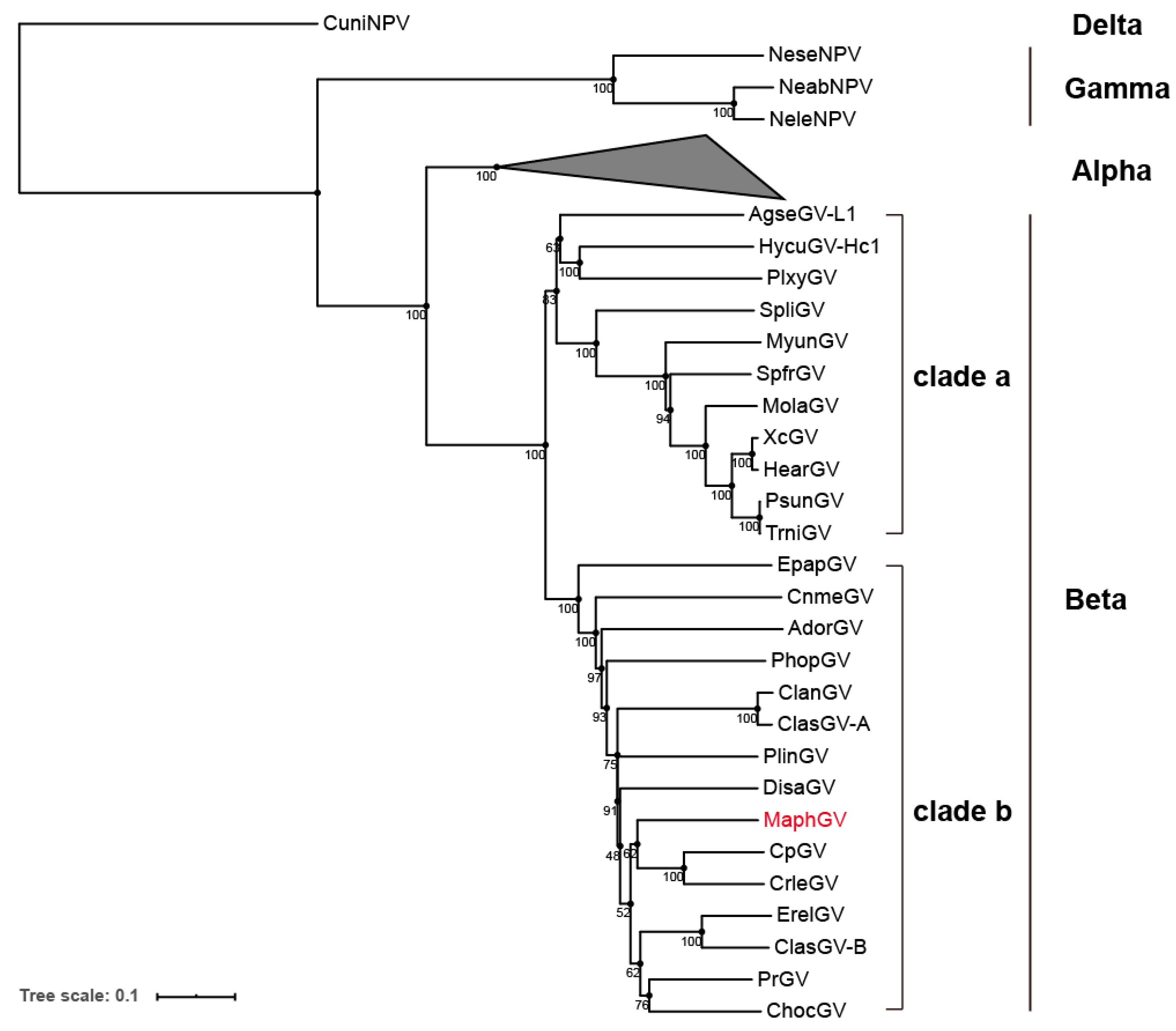
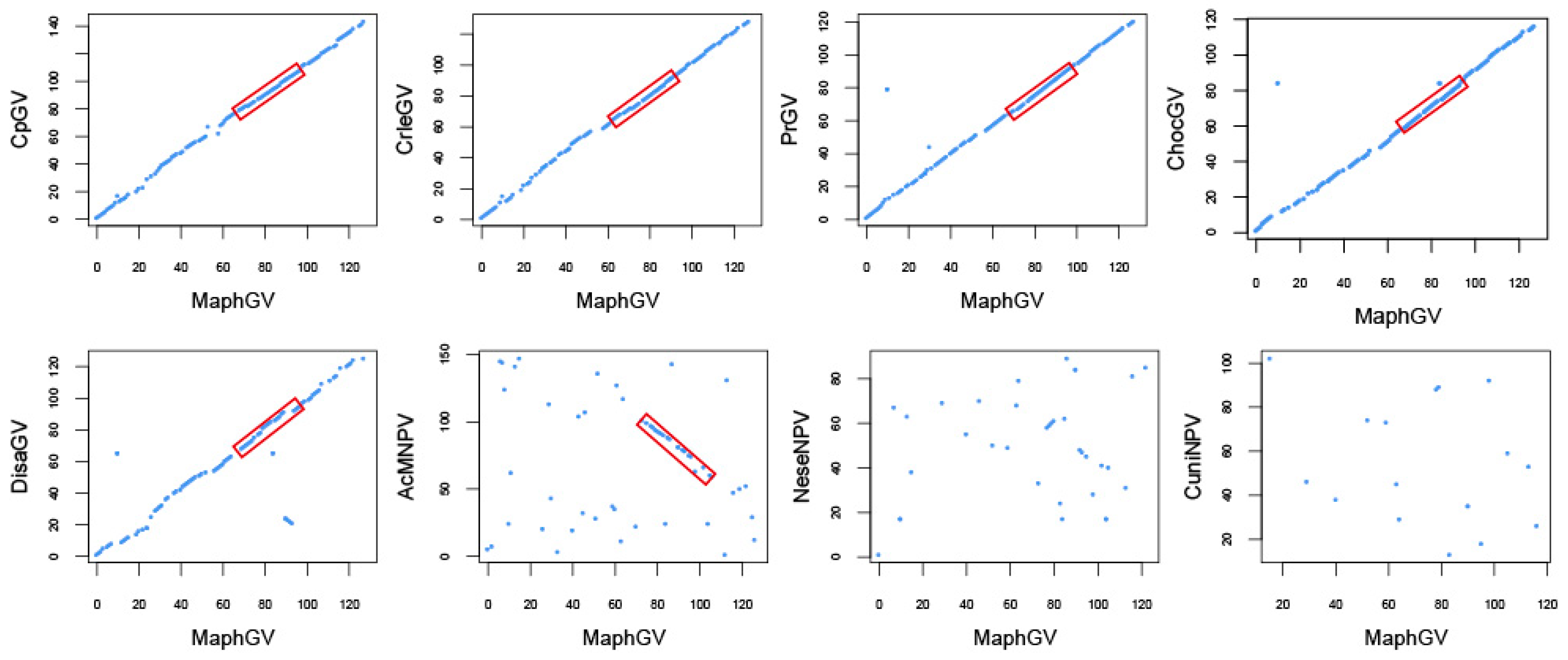
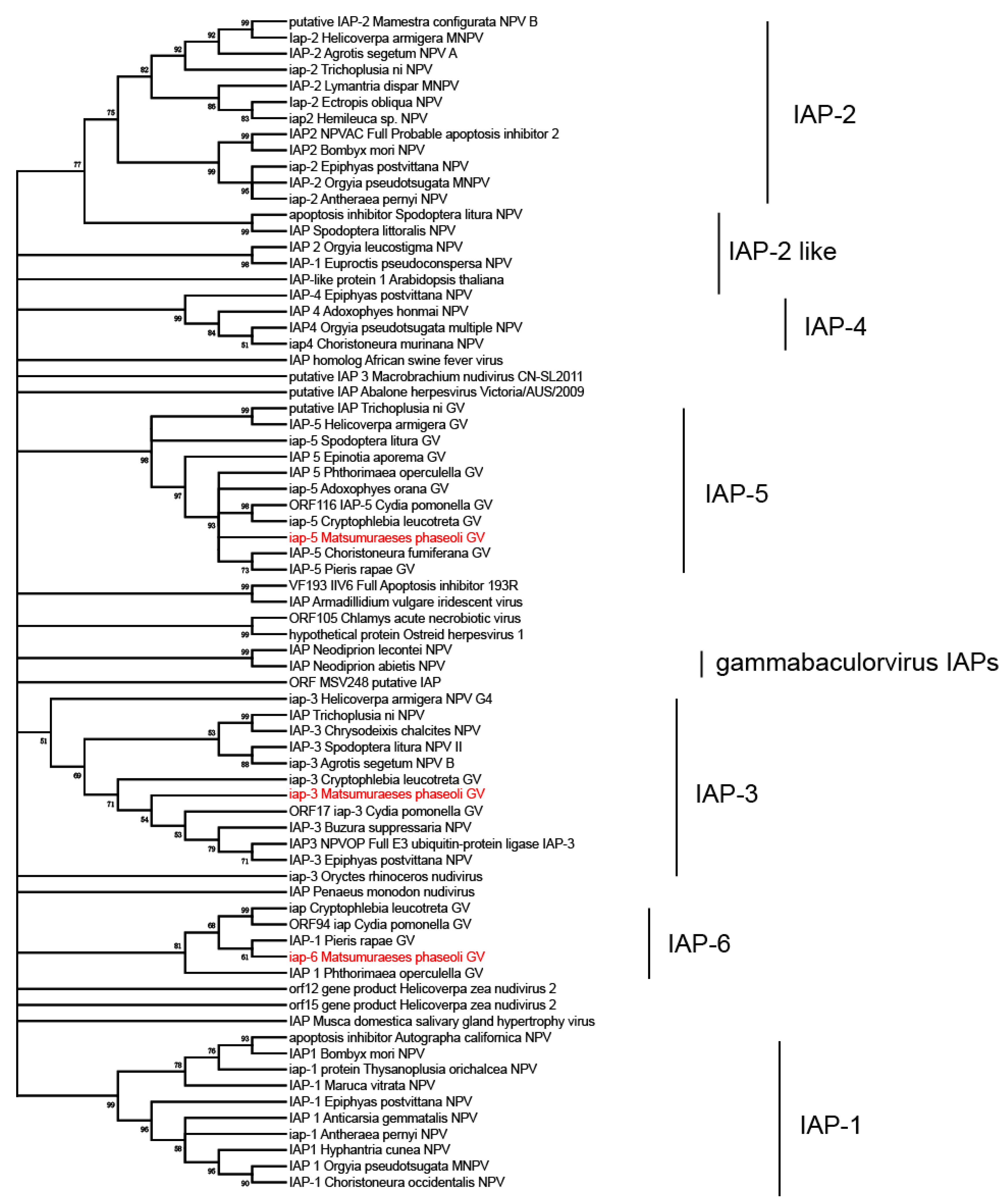
3.3. Phylogenetic Analysis of MaphGV
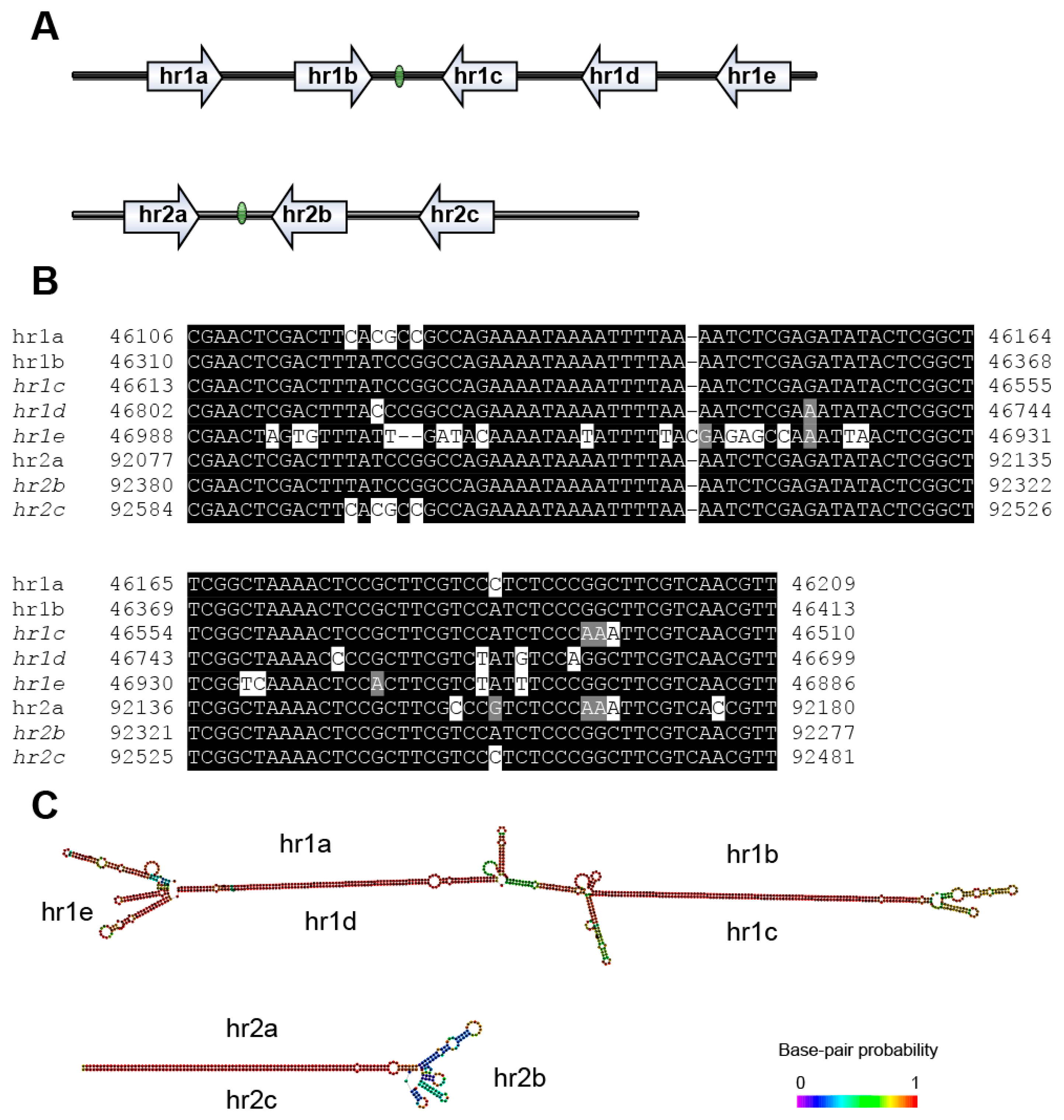
3.4. Repeat Sequences
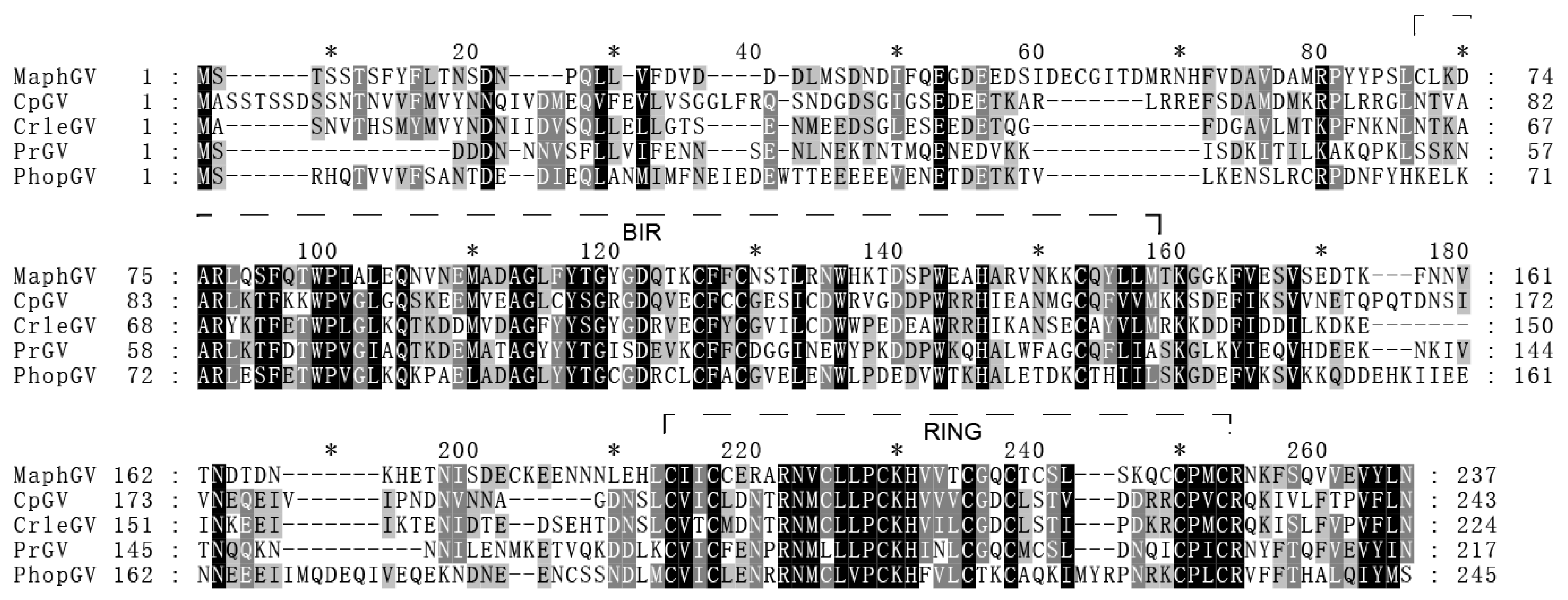
3.5. iap-6 Found in MaphGV Genome
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Harrison, R.L.; Herniou, E.A.; Jehle, J.A.; Theilmann, D.A.; Burand, J.P.; Becnel, J.J.; Krell, P.J.; van Oers, M.M.; Mowery, J.D.; Bauchan, G.R.; et al. ICTV Virus Taxonomy Profile: Baculoviridae. J. Gen. Virol. 2018, 99, 1185–1186. [Google Scholar] [CrossRef]
- Kelly, B.J.; King, L.A.; Possee, R.D. Introduction to Baculovirus Molecular Biology. In Baculovirus and Insect Cell Expression Protocols; Murhammer, D.W., Ed.; Methods in Molecular Biology; Springer: New York, NY, USA, 2016; pp. 25–50. ISBN 978-1-4939-3043-2. [Google Scholar]
- Jehle, J.A.; Blissard, G.W.; Bonning, B.C.; Cory, J.S.; Herniou, E.A.; Rohrmann, G.F.; Theilmann, D.A.; Thiem, S.M.; Vlak, J.M. On the classification and nomenclature of baculoviruses: A proposal for revision. Arch. Virol. 2006, 151, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- Clem, R.J.; Passarelli, A.L. Baculoviruses: Sophisticated Pathogens of Insects. PLoS Pathog. 2013, 9, e1003729. [Google Scholar] [CrossRef]
- Herniou, E.A.; Olszewski, J.A.; O’Reilly, D.R.; Cory, J.S. Ancient Coevolution of Baculoviruses and Their Insect Hosts. J. Virol. 2004, 78, 3244–3251. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, G.F. Baculovirus Molecular Biology, 4th ed.; National Center for Biotechnology Information (US): Bethesda, MD, USA, 2019.
- Capinera, J.L. (Ed.) Encyclopedia of Entomology, 2nd ed.; Springer reference; Springer Science+Business Media B.V: Dordrecht, The Netherlands, 2008; ISBN 978-1-4020-6242-1. [Google Scholar]
- Liu, P.; Zhao, H.; Luo, Y. Anti-Aging Implications of Astragalus Membranaceus (Huangqi): A Well-Known Chinese Tonic. Aging Dis. 2017, 8, 868–886. [Google Scholar] [CrossRef] [PubMed]
- Byun, B.-K.; Park, K.-T.; Park, Y.-M. Review of the Genus Matsumuraeses Issiki (Lepidoptera, Tortricidae) with Discovery of M. falcana (Walsingham) in Korea. J. Asia-Pac. Entomol. 2005, 8, 117–122. [Google Scholar] [CrossRef]
- Lü, H. Preliminary studies on the soy-bean leaf-roller Matsumuraeses phaseoli (Mats.), (Lepidoptera: Olethreutidae) Ⅰ. morphology and biological characteristics. Acta Phytophylacica Sin. 1965, 4, 255–269. (In Chinese) [Google Scholar] [CrossRef]
- Martignoni, M.E. A Catalog of Viral Diseases of Insects, Mites, and Ticks; U.S. Department of Agriculture, Forest Service, Pacific Northwest Research Station: Corvallis, OR, USA, 1986.
- Hull, R.; Brown, F.; Payne, C. Virology: A Directory and Dictionary of Animal, Bacterial and Plant Viruses; Springer: Berlin, Germany, 1989; ISBN 978-1-349-07945-2. [Google Scholar]
- Hajibabaei, M.; Janzen, D.H.; Burns, J.M.; Hallwachs, W.; Hebert, P.D.N. DNA barcodes distinguish species of tropical Lepidoptera. Proc. Natl. Acad. Sci. USA 2006, 103, 968–971. [Google Scholar] [CrossRef]
- Pinto, F.A.; Mattos, M.V.V.; Silva, F.W.S.; Rocha, S.L.; Elliot, S.L. The Spread of Helicoverpa armigera (Lepidoptera: Noctuidae) and Coexistence with Helicoverpa zea in Southeastern Brazil. Insects 2017, 8, 87. [Google Scholar] [CrossRef]
- Ratnasingham, S.; Hebert, P.D.N. BLOD: The Barcode of Life Data System (http://www.barcodinglife.org). Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef]
- Seo, B.Y.; Jung, J.K.; Cho, J.R.; Kim, Y.; Park, C.G. A PCR method to distinguish Matsumuraeses phaseoli from M falcana based on the difference of nucleotide sequence in the mitochondrial cytochrome c oxidase subunit I. Korean J. Appl. Entomol. 2012, 51, 365–370. [Google Scholar] [CrossRef]
- Tang, P.; Zhang, H.; Li, Y.; Han, B.; Wang, G.; Qin, Q.; Zhang, Z. Genomic sequencing and analyses of HearMNPV—a new Multinucleocapsid nucleopolyhedrovirus isolated from Helicoverpa armigera. Virol. J. 2012, 9, 168. [Google Scholar] [CrossRef] [PubMed]
- Ardisson-Araújo, D.M.P.; de Melo, F.L.; de Souza Andrade, M.; Sihler, W.; Báo, S.N.; Ribeiro, B.M.; de Souza, M.L. Genome sequence of Erinnyis ello granulovirus (ErelGV), a natural cassava hornworm pesticide and the first sequenced sphingid-infecting betabaculovirus. BMC Genomics 2014, 15, 856. [Google Scholar] [CrossRef]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; ISBN 978-0-87969-577-4. [Google Scholar]
- Hunt, M.; Gall, A.; Ong, S.H.; Brener, J.; Ferns, B.; Goulder, P.; Nastouli, E.; Keane, J.A.; Kellam, P.; Otto, T.D. IVA: Accurate de novo assembly of RNA virus genomes. Bioinformatics 2015, 31, 2374–2376. [Google Scholar] [CrossRef] [PubMed]
- Wennmann, J.T.; Fan, J.; Jehle, J.A. Bacsnp: Using Single Nucleotide Polymorphism (SNP) Specificities and Frequencies to Identify Genotype Composition in Baculoviruses. Viruses 2020, 12, 625. [Google Scholar] [CrossRef]
- Solovyev, V.; Kosarev, P.; Seledsov, I.; Vorobyev, D. Automatic annotation of eukaryotic genes, pseudogenes and promoters. Genome Biol. 2006, 7, S10. [Google Scholar] [CrossRef]
- Wang, S.; Sundaram, J.P.; Spiro, D. VIGOR, an annotation program for small viral genomes. BMC Bioinformatics 2010, 11, 451. [Google Scholar] [CrossRef]
- Madeira, F.; Park, Y.M.; Lee, J.; Buso, N.; Gur, T.; Madhusoodanan, N.; Basutkar, P.; Tivey, A.R.N.; Potter, S.C.; Finn, R.D.; et al. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res. 2019, 47, W636–W641. [Google Scholar] [CrossRef]
- Hu, Z.H.; Arif, B.M.; Jin, F.; Martens, J.W.M.; Chen, X.W.; Sun, J.S.; Zuidema, D.; Goldbach, R.W.; Vlak, J.M. Distinct gene arrangement in the Buzura suppressaria single-nucleocapsid nucleopolyhedrovirus genome. J. Gen. Virol. 1998, 79, 2841–2851. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Lorenz, R.; Bernhart, S.H.; Höner zu Siederdissen, C.; Tafer, H.; Flamm, C.; Stadler, P.F.; Hofacker, I.L. ViennaRNA Package 2.0. Algorithms Mol. Biol. 2011, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Xie, Y.; Ma, J.; Luo, X.; Nie, P.; Zuo, Z.; Lahrmann, U.; Zhao, Q.; Zheng, Y.; Zhao, Y.; et al. IBS: An illustrator for the presentation and visualization of biological sequences. Bioinformatics 2015, 31, 3359–3361. [Google Scholar] [CrossRef] [PubMed]
- BoxShade. Available online: https://embnet.vital-it.ch/software/BOX_form.html (accessed on 15 July 2020).
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Guarino, L.A.; Smith, M. Regulation of delayed-early gene transcription by dual TATA boxes. J. Virol. 1992, 66, 3733–3739. [Google Scholar] [CrossRef]
- Blissard, G.W.; Kogan, P.H.; Wei, R.; Rohrmann, G.F. A synthetic early promoter from a baculovirus: Roles of the TATA box and conserved start site CAGT sequence in basal levels of transcription. Virology 1992, 190, 783–793. [Google Scholar] [CrossRef]
- Wormleaton, S.; Kuzio, J.; Winstanley, D. The complete sequence of the Adoxophyes orana granulovirus genome. Virology 2003, 311, 350–365. [Google Scholar] [CrossRef]
- Morris, T.D.; Miller, L.K. Mutational analysis of a baculovirus major late promoter. Gene 1994, 140, 147–153. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Criscuolo, A.; Gribaldo, S. BMGE (Block Mapping and Gathering with Entropy): A new software for selection of phylogenetic informative regions from multiple sequence alignments. BMC Evol. Biol. 2010, 10, 210. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Paradis, E.; Claude, J.; Strimmer, K. APE: Analyses of Phylogenetics and Evolution in R language. Bioinforma. Oxf. Engl. 2004, 20, 289–290. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- King, A.M.Q.; Adams, M.J. Virus Taxonomy Ninth Report of the International Committee on Taxonomy of Viruses; Elsevier Science & Technology Books: San Diego, CA, USA, 2011; ISBN 978-0-12-384685-3. [Google Scholar]
- Garavaglia, M.J.; Miele, S.A.B.; Iserte, J.A.; Belaich, M.N.; Ghiringhelli, P.D. The ac53, ac78, ac101, and ac103 Genes Are Newly Discovered Core Genes in the Family Baculoviridae. J. Virol. 2012, 86, 12069–12079. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Lange, M.; Wang, H.; Hu, Z.; Wang, Y.; Hauschild, R. Molecular identification and phylogenetic analysis of baculoviruses from Lepidoptera. Virology 2006, 346, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Wennmann, J.T.; Keilwagen, J.; Jehle, J.A. Baculovirus Kimura two-parameter species demarcation criterion is confirmed by the distances of 38 core gene nucleotide sequences. J. Gen. Virol. 2018, 99, 1307–1320. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.A.; Olszewski, J.A.; Cory, J.S.; O’Reilly, D.R. The genome sequence and evolution of baculoviruses. Annu. Rev. Entomol. 2003, 48, 211–234. [Google Scholar] [CrossRef] [PubMed]
- Hilton, S.; Winstanley, D. The origins of replication of granuloviruses. Arch. Virol. 2008, 153, 1527–1535. [Google Scholar] [CrossRef] [PubMed]
- Harrison, R.L.; Rowley, D.L.; Mowery, J.; Bauchan, G.R.; Theilmann, D.A.; Rohrmann, G.F.; Erlandson, M.A. The Complete Genome Sequence of a Second Distinct Betabaculovirus from the True Armyworm, Mythimna unipuncta. PLoS ONE 2017, 12, e0170510. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, T.; Ko, R.; Okano, K.; Seong, S.I.; Goto, C.; Maeda, S. Sequence analysis of the Xestia c-nigrum granulovirus genome. Virology 1999, 262, 277–297. [Google Scholar] [CrossRef] [PubMed]
- Cuartas, P.E.; Barrera, G.P.; Belaich, M.N.; Barreto, E.; Ghiringhelli, P.D.; Villamizar, L.F. The Complete Sequence of the First Spodoptera frugiperda Betabaculovirus Genome: A Natural Multiple Recombinant Virus. Viruses 2015, 7, 394–421. [Google Scholar] [CrossRef]
- Kang, W.; Suzuki, M.; Zemskov, E.; Okano, K.; Maeda, S. Characterization of Baculovirus Repeated Open Reading Frames (bro) in Bombyx mori Nucleopolyhedrovirus. J. Virol. 1999, 73, 10339–10345. [Google Scholar] [CrossRef]
- Clem, R.J. Viral IAPs, then and now. Semin. Cell Dev. Biol. 2015, 39, 72–79. [Google Scholar] [CrossRef]
- Harrison, R.L.; Rowley, D.L.; Funk, C.J. The Complete Genome Sequence of Plodia Interpunctella Granulovirus: Evidence for Horizontal Gene Transfer and Discovery of an Unusual Inhibitor-of-Apoptosis Gene. PLoS ONE 2016, 11, e0160389. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Type | Core Genes | Lepidoptera Baculovirus Conserved Genes | Other Baculovirus Genes |
|---|---|---|---|
| Structure | odv-e18 (orf13), 49k (orf14), odv-ec43 (orf47), p48/p45 (orf74), p40;bv/odv-c42 (orf76), p6.9 (orf77), 38k (orf79), odv-e25 (orf82), p18 (orf83), p33 (orf84), vp39 (orf87), odv-ec27 (orf88), ac81 (orf93), gp41 (orf94), ac78 (orf95), desmop (orf100), ac53 (orf120), vp1054 (orf123) | granulin (orf1), pk-1 (orf3), calyx/pep-1 (orf20), F protein (orf27), bv-e31 (orf61), p24 (orf62), p12 (orf75), tlp-20 (orf92), fp25k (orf118) | p10 (orf17), pep-2 (orf23), vp80 (orf24) |
| Replication | lef-2 (orf34), lef-1 (orf64), helicase (orf81), dna-pol (orf99), alk-exo (orf114) | lef-11 (orf51), dbp (orf71), lef-3 (orf102), me53 (orf128) | ie-1 (orf6), dna ligase (orf108), helicase-2 (orf115) |
| Transcription | p47 (orf60), lef-5 (orf78), lef-4 (orf86), vlf1 (orf96), lef-9 (orf106), lef-8 (orf117) | 39k (orf50), lef-6 (orf70) | |
| Oral infection | pif-5 (orf16), pif-3 (orf30), pif-2 (orf41), ac110 (orf45), p74 (orf53), pif-1 (orf65), pif-4 (orf80), pif-8 (orf91), pif-6 (orf103) | odv-e66 (orf31) | |
| Auxiliary gene | 38.7k (orf63) | chitinase (orf9), iap-3 (orf11), gp37 (orf12), bro (orf26), p13 (orf39), ubiquitin (orf46), sod (orf52), fgf-1 (orf66), iap-1 (orf85), iap-2 (orf105), fgf-2 (orf111), fgf-3 (orf126), egt (orf127) | |
| Unique gene | orf22, orf40, orf56, orf101, orf124 | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shu, R.; Meng, Q.; Miao, L.; Liang, H.; Chen, J.; Xu, Y.; Cheng, L.; Jin, W.; Qin, Q.; Zhang, H. Genome Analysis of a Novel Clade b Betabaculovirus Isolated from the Legume Pest Matsumuraeses phaseoli (Lepidoptera: Tortricidae). Viruses 2020, 12, 1068. https://doi.org/10.3390/v12101068
Shu R, Meng Q, Miao L, Liang H, Chen J, Xu Y, Cheng L, Jin W, Qin Q, Zhang H. Genome Analysis of a Novel Clade b Betabaculovirus Isolated from the Legume Pest Matsumuraeses phaseoli (Lepidoptera: Tortricidae). Viruses. 2020; 12(10):1068. https://doi.org/10.3390/v12101068
Chicago/Turabian StyleShu, Ruihao, Qian Meng, Lin Miao, Hongbin Liang, Jun Chen, Yuan Xu, Luqiang Cheng, Wenyi Jin, Qilian Qin, and Huan Zhang. 2020. "Genome Analysis of a Novel Clade b Betabaculovirus Isolated from the Legume Pest Matsumuraeses phaseoli (Lepidoptera: Tortricidae)" Viruses 12, no. 10: 1068. https://doi.org/10.3390/v12101068
APA StyleShu, R., Meng, Q., Miao, L., Liang, H., Chen, J., Xu, Y., Cheng, L., Jin, W., Qin, Q., & Zhang, H. (2020). Genome Analysis of a Novel Clade b Betabaculovirus Isolated from the Legume Pest Matsumuraeses phaseoli (Lepidoptera: Tortricidae). Viruses, 12(10), 1068. https://doi.org/10.3390/v12101068