Depicting the RNA Virome of Hematophagous Arthropods from Belgrade, Serbia

 ,
,  , and
, and
Abstract
1. Introduction
2. Methods
2.1. Collection and Processing of Arthropods
2.2. Sample Processing and RNA Extraction
2.3. RNA-Seq Sequencing, Reads Assembly, Virus Sequences Discovery and Confirmation
2.4. Acquisition of the Coding-Complete Sequences or RdRp Genes
2.5. Phylogenetic Analysis
3. Results
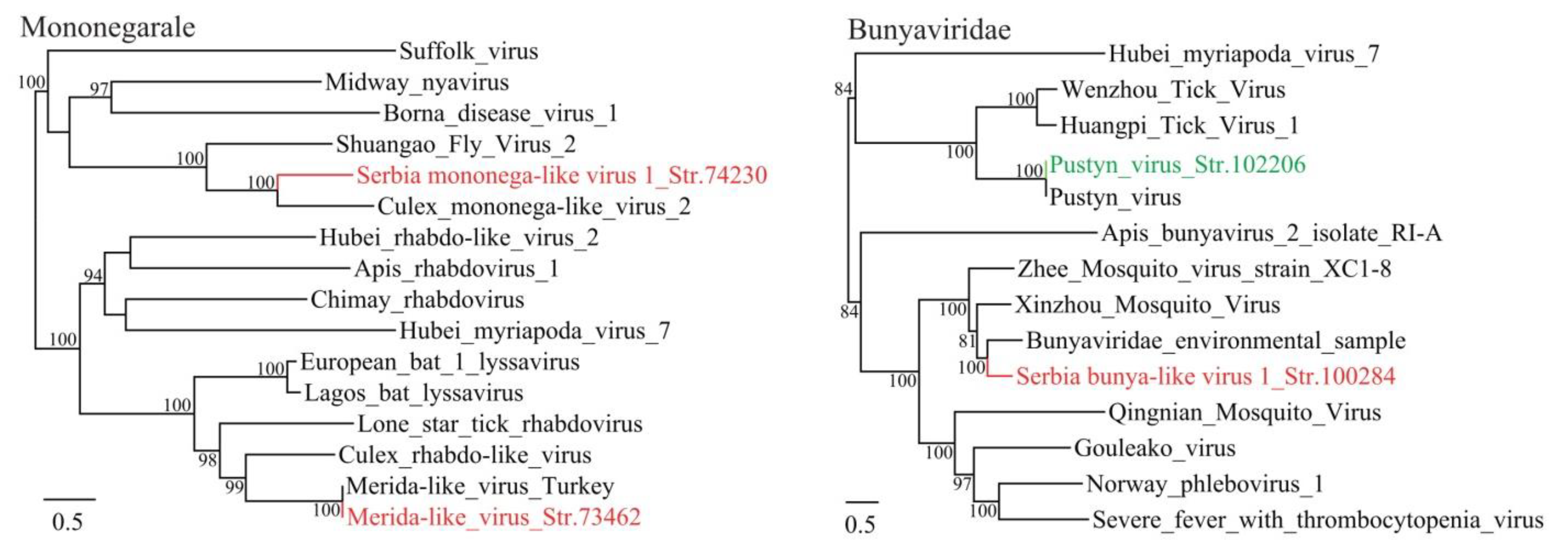
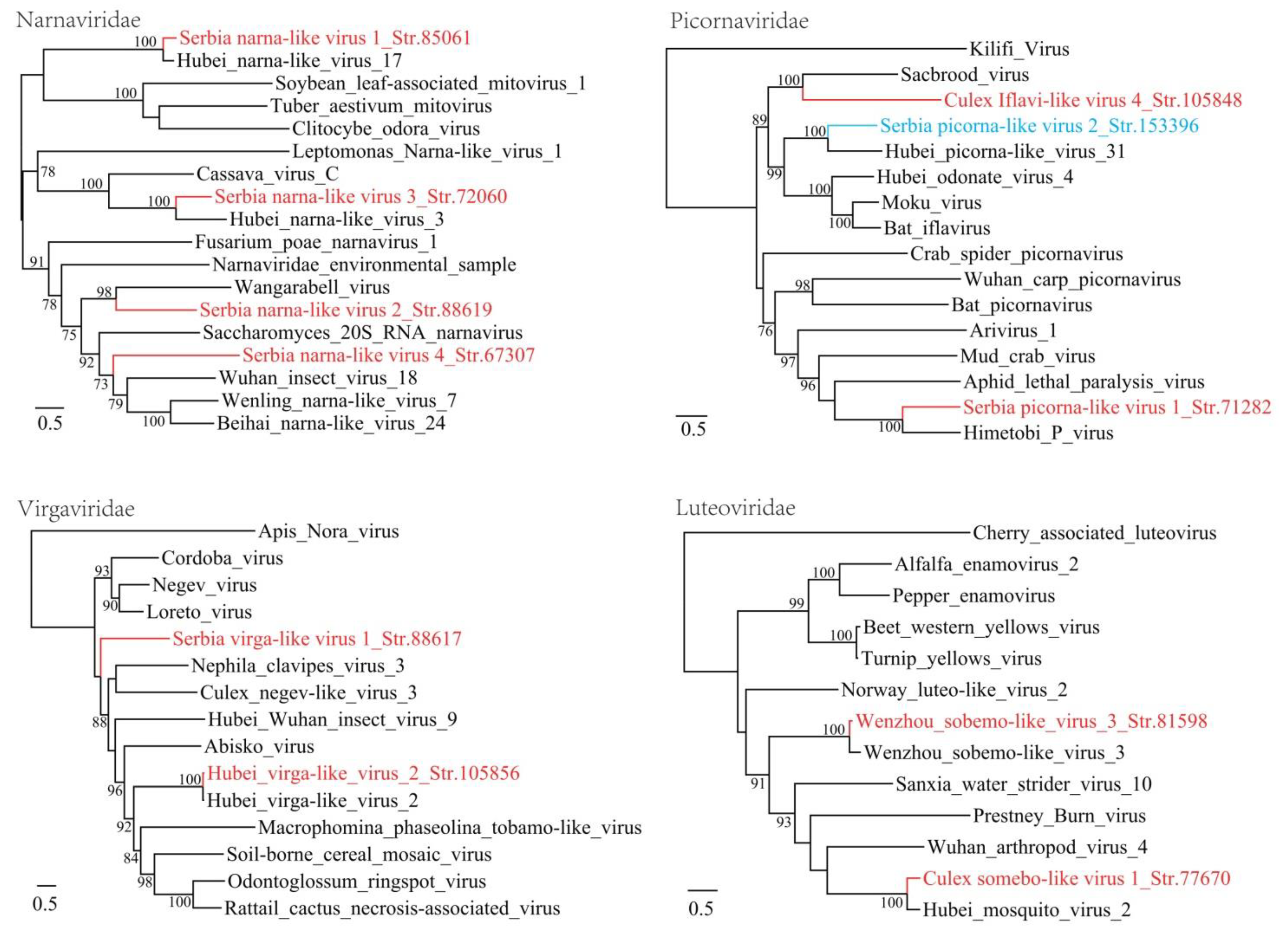
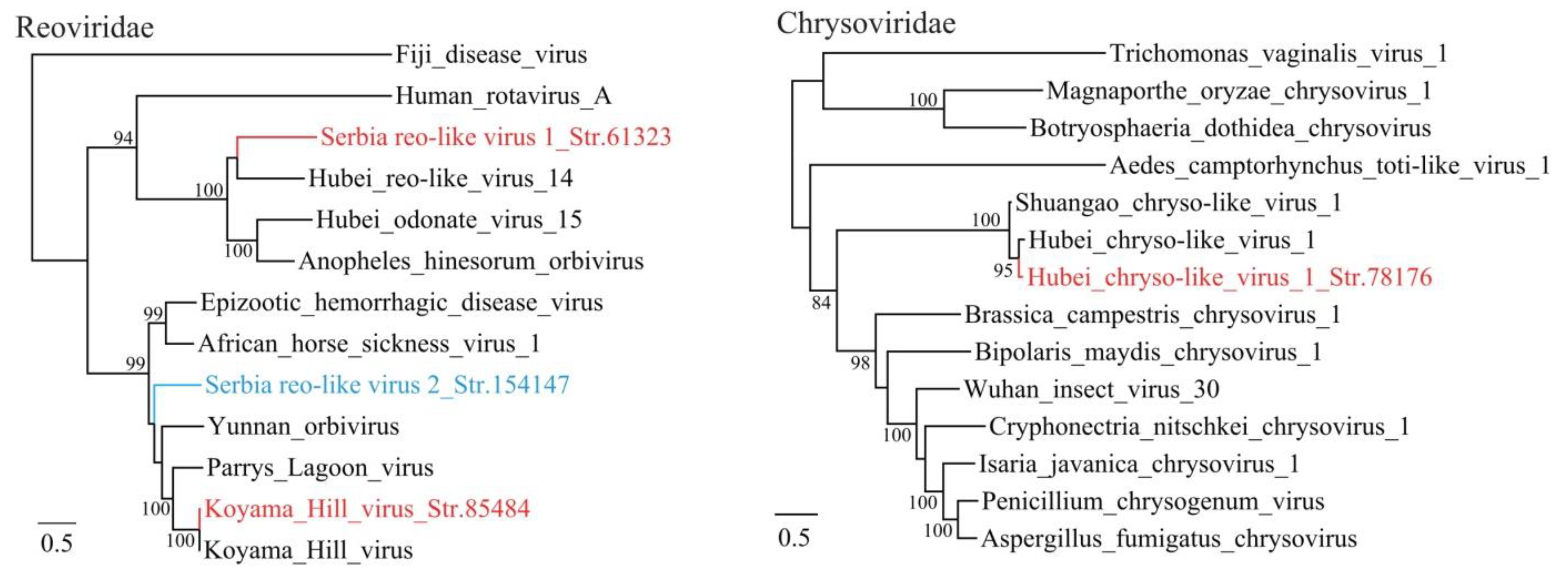
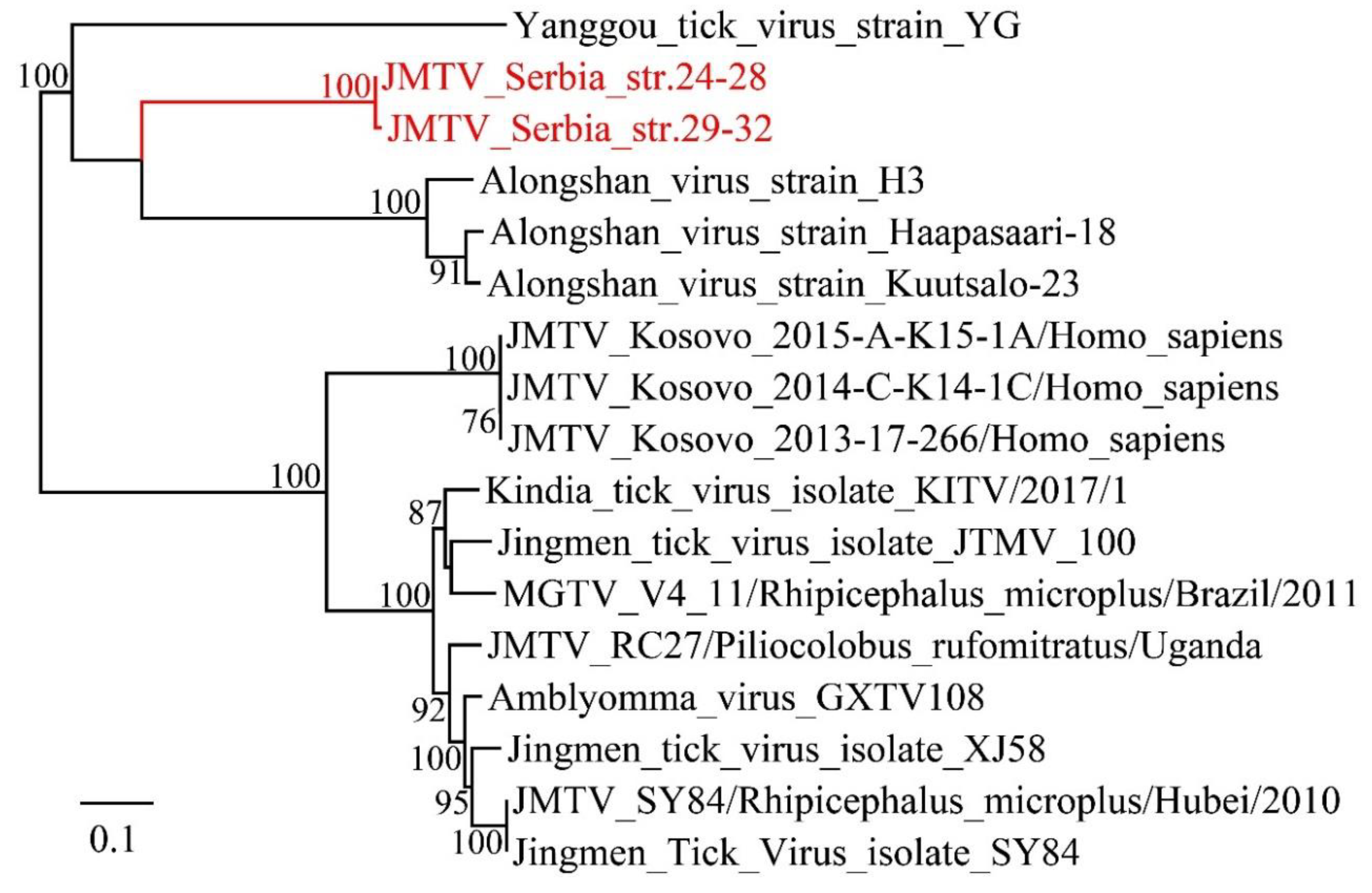
3.1. Discovery of Divergent RNA Viruses in Arthropods
3.2. Diverse Viruses Identified in Mosquitoes
3.3. Viruses Identified in Ticks, Bedbugs and Fleas
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Whitman, J.D.; Yanega, D.; Watson, C.B.G.; Strode, V.W. Collection and preservation of terrestrial arthropods. Methods Mol. Biol. 2019, 1897, 163–189. [Google Scholar]
- Tabachnick, W.J. Ecological effects on arbovirus-mosquito cycles of transmission. Curr. Opin. Virol. 2016, 21, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.D.; Tian, J.H.; Chen, L.J.; Chen, X.; Li, C.X.; Qin, X.C.; Li, J.; Cao, J.P.; Eden, J.S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Li, C.X.; Shi, M.; Tian, J.H.; Lin, X.D.; Kang, Y.J.; Chen, L.J.; Qin, X.C.; Xu, J.; Holmes, E.C.; Zhang, Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4, e05378. [Google Scholar] [CrossRef]
- Cutler, S.J.; Fooks, A.R.; van der Poel, W.H. Public health threat of new, reemerging, and neglected zoonoses in the industrialized world. Emerg. Infect Dis. 2010, 16, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.C.; Reisen, W.K. Present and future arboviral. Threat. Antivir. Res. 2010, 85, 328–345. [Google Scholar] [CrossRef]
- Liang, G.; Li, X.; Gao, X.; Fu, S.; Wang, H.; Li, M.; Lu, Z.; Zhu, W.; Lu, X.; Wang, L.; et al. Arboviruses and their related infections in China: A comprehensive field and laboratory investigation over the last 3 decades. Rev. Med. Virol. 2018, 28. [Google Scholar] [CrossRef]
- Calisher, C.H.; Higgs, S. The discovery of arthropod-specific viruses in hematophagous arthropods: An open door to understanding the mechanisms of arbovirus and arthropod evolution? Annu. Rev. Entomol. 2018, 63, 87–103. [Google Scholar] [CrossRef]
- Tomori, O. Yellow fever: The recurring plague. Crit. Rev. Clin. Lab. Sci. 2004, 41, 391–427. [Google Scholar] [CrossRef]
- Byard, R.W. Lethal dengue virus infection: A forensic overview. Am. J. Forensic. Med. Pathol. 2016, 37, 74–78. [Google Scholar] [CrossRef]
- Atoni, E.; Wang, Y.; Karungu, S.; Waruhiu, C.; Zohaib, A.; Obanda, V.; Agwanda, B.; Mutua, M.; Xia, H.; Yuan, Z. Metagenomic virome analysis of culex mosquitoes from Kenya and China. Viruses 2018, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Hu, C.; Zhang, D.; Tang, S.; Zhang, Z.; Kou, Z.; Fan, Z.; Bente, D.; Zeng, C.; Li, T. Metagenomic profile of the viral communities in Rhipicephalus spp. ticks from Yunnan, China. PLoS ONE 2015, 10, e0121609. [Google Scholar] [CrossRef]
- Harvey, E.; Rose, K.; Eden, J.S.; Lo, N.; Abeyasuriya, T.; Shi, M.; Doggett, S.L.; Holmes, E.C. Extensive diversity of RNA viruses in Australian ticks. J. Virol. 2019, 93, e01358:1–e01358:18. [Google Scholar] [CrossRef] [PubMed]
- Sadeghi, M.; Altan, E.; Deng, X.; Barker, C.M.; Fang, Y.; Coffey, L.L.; Delwart, E. Virome of > 12 thousand Culex mosquitoes from throughout California. Virology 2018, 523, 74–88. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, J.H.; Shi, M.; Bohlin, J.; Eldholm, V.; Brynildsrud, O.B.; Paulsen, K.M.; Andreassen, Å.; Holmes, E.C. Characterizing the virome of Ixodes ricinus ticks from northern Europe. Sci. Rep. 2017, 7, 10870. [Google Scholar] [CrossRef]
- Temmam, S.; Monteil-Bouchard, S.; Robert, C.; Baudoin, J.P.; Sambou, M.; Aubadie-Ladrix, M.; Labas, N.; Raoult, D.; Mediannikov, O.; Desnues, C. Characterization of viral communities of biting midges and identification of novel thogotovirus species and rhabdovirus genus. Viruses 2016, 8, 77. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Ergünay, K.; Brinkmann, A.; Litzba, N.; Günay, F.; Kar, S.; Öter, K.; Örsten, S.; Sarıkaya, Y.; Alten, B.; Nitsche, A.; et al. A novel rhabdovirus, related to Merida virus, in field-collected mosquitoes from Anatolia and Thrace. Arch. Virol. 2017, 162, 1903–1911. [Google Scholar] [CrossRef]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.S.; Imrie, A.; Holmes, E.C. High-resolution metatranscriptomics reveals the ecological dynamics of mosquito-associated RNA viruses in Western Australia. J. Virol. 2017, 91, e00680-17. [Google Scholar] [CrossRef]
- Adams, M.J.; Adkins, S.; Bragard, C.; Gilmer, D.; Li, D.; MacFarlane, S.A.; Wong, S.M.; Melcher, U.; Ratti, C.; Ryu, K.H. Ictv report consortium. ICTV virus taxonomy profile: Virgaviridae. J. Gen. Virol. 2017, 98, 1999–2000. [Google Scholar] [CrossRef] [PubMed]
- Nunes, M.R.T.; Contreras-Gutierrez, M.A.; Guzman, H.; Martins, L.C.; Barbirato, M.F.; Savit, C.; Balta, V.; Uribe, S.; Vivero, R.; Suaza, J.D.; et al. Genetic characterization, molecular epidemiology, and phylogenetic relationships of insect-specific viruses in the taxon Negevirus. Virology 2017, 504, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Hillman, B.I.; Cai, G. The family narnaviridae: Simplest of RNA viruses. Adv. Virus. Res. 2013, 86, 149–176. [Google Scholar] [PubMed]
- Chandler, J.A.; Liu, R.M.; Bennett, S.N. RNA shotgun metagenomic sequencing of northern California (USA) mosquitoes uncovers viruses, bacteria, and fungi. Fron. Microbiol. 2015, 6, 185. [Google Scholar] [CrossRef]
- Toriyama, S.; Guy, P.L.; Fuji, S.; Takahashi, M. Characterization of a new picorna-like virus, himetobi P virus, in planthoppers. J. Gen. Virol. 1992, 73, 1021–1023. [Google Scholar] [CrossRef]
- Boros, Á.; Pankovics, P.; Simmonds, P.; Pollák, E.; Mátics, R.; Phan, T.G.; Delwart, E.; Reuter, G. Genome analysis of a novel, highly divergent picornavirus from common kestrel (Falco tinnunculus): The first non-enteroviral picornavirus with type-I-like IRES. Infect. Genet. Evol. 2015, 32, 425–431. [Google Scholar] [CrossRef]
- Temmam, S.; Bigot, T.; Chrétien, D.; Gondard, M.; Pérot, P.; Pommelet, V.; Dufour, E.; Petres, S.; Devillers, E.; Hoem, T.; et al. Insights into the host range, genetic diversity, and geographical distribution of jingmenviruses. MSphere. 2019, 4, e00645-19. [Google Scholar] [CrossRef]
- Wang, Z.D.; Wang, B.; Wei, F.; Han, S.Z.; Zhang, L.; Yang, Z.T.; Yan, Y.; Lv, X.L.; Li, L.; Wang, S.C.; et al. A new segmented virus associated with human febrile illness in China. N. Engl. J. Med. 2019, 380, 2116–2125. [Google Scholar] [CrossRef]
- Emmerich, P.; Jakupi, X.; von Possel, R.; Berisha, L.; Halili, B.; Günther, S.; Cadar, D.; Ahmeti, S.; Schmidt-Chanasit, J. Viral metagenomics, genetic and evolutionary characteristics of Crimean-Congo hemorrhagic fever orthonairovirus in humans, Kosovo. Infect Genet. Evol. 2018, 65, 6–11. [Google Scholar] [CrossRef]
- Harrison, J.J.; Warrilow, D.; McLean, B.J.; Watterson, D.; O’Brien, C.A.; Colmant, A.M.; Johansen, C.A.; Barnard, R.T.; Hall-Mendelin, S.; Davis, S.S.; et al. A new Orbivirus isolated from mosquitoes in North-Western Australia shows antigenic and genetic similarity to Corriparta Virus but does not replicate in vertebrate cells. Viruses 2016, 8, 141. [Google Scholar] [CrossRef]
- Maclachlan, N.J.; Guthrie, A.J. Re-emergence of bluetongue, African horse sickness, and other orbivirus diseases. Vet. Res. 2010, 41, 35. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, H.; Sun, X.; Fu, S.; Wang, H.; Feng, Y.; Wang, H.; Tang, Q.; Liang, G.D. Distribution of mosquitoes and mosquito-borne arboviruses in Yunnan province near the China–Myanmar–Laos border. Am. J. Trop. Med. Hyg. 2011, 84, 738–746. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Stanojević, M.; Stamenković, G.; Ilić, B.; Paunović, M.; Lu, M.; Pešić, B.; Đurić Maslovara, I.; Siljic, M.; Cirkovic, V.; et al. Insight into diversity of bacteria belonging to the order rickettsiales in 9 arthropods species collected in Serbia. Sci. Rep. 2019, 9, 18680. [Google Scholar] [CrossRef]
- Gligic, A.; Stamatovic, I.; Stojanovic, R.; Obradovic, M.; Boskovic, R. The first isolation of the crimean hemorraghic fever virus in Yugoslavia. Vojn. Pregl. 1977, 34, 318–321. [Google Scholar]
- Popović, N.; Milošević, B.; Urošević, A.; Poluga, J.; Lavadinović, L.; Nedelijković, J.; Jevtović, D.; Dulović, O. Outbreak of west nile virus infection among humans in Serbia, august to october 2012. Eur. Surveill. 2013, 18. [Google Scholar] [CrossRef]
- Petrović, T.; Šekler, M.; Petrić, D.; Lazić, S.; Debeljak, Z.; Vidanović, D.; Ignjatović Ćupina, A.; Lazić, G.; Lupulović, D.; Kolarević, M.; et al. Methodology and results of integrated WNV surveillance programmes in Serbia. PLoS ONE 2018, 13, e0195439. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Location | Coordinates | Species | Pool Size | No. of Sequenced Reads | Average Length of Reads |
|---|---|---|---|---|---|
| Belgrade | N44.759–N44.881 E20.244–E20.625 | Culex pipiens L | 360 | 43699119 | 150 |
| Belgrade and surrounding areas | N42.710–N45.455 E19.220–E22.342 | Ixodes ricinus | 37 | 28141701 | 150 |
| Belgrade and surrounding areas | N44.690–N44.935 E20.450–E21.136 | Cimex lectularius and other arthropods | 118 | 34233162 | 150 |
| Provisional Name | Accession Number | Host | Taxon | Best Hit | Coverage (%) | Identity (%) | Sequence Length | Obtained Region |
|---|---|---|---|---|---|---|---|---|
| Serbia mononega-like virus 1_Str. 74230 | MT822181 | Culex pipiens | Mononegavirale | Culex mononega-like virus 2 | 43 | 41 | 12,933 | Complete CDS |
| Serbia bunya-like virus 1_Str. 100284 | MT822182 | Culex pipiens | Bunyavirales | Bunyaviridae environmental sample | 95 | 57 | 7649 | Complete RdRp |
| Serbia narna-like virus 1_Str. 85061 | MT822183 | Culex pipiens | Narnaviridae | Hubei narna-like virus 17 | 83 | 71 | 3280 | Complete RdRp |
| Serbia narna-like virus 2_Str. 88619 | MT822184 | Culex pipiens | Narnaviridae | Wangarabell virus | 90 | 30 | 5269 | Complete RdRp |
| Serbia narna-like virus 3_Str. 72060 | MT822185 | Culex pipiens | Narnaviridae | Hubei narna-like virus 3 | 89 | 48 | 2685 | Complete RdRp |
| Serbia narna-like virus 4_Str. 67307 | MT822186 | Culex pipiens | Narnaviridae | Hubei narna-like virus 16 | 70 | 44 | 2889 | Complete RdRp |
| Serbia picorna-like virus 1_Str. 71282 | MT822187 | Culex pipiens | Picornavirale | Himetobi P virus | 56 | 41 | 9132 | Complete CDS |
| Serbia virga-like virus 1_Str. 88617 | MT822191 | Culex pipiens | Virgaviridae | Loreto virus | 43 | 31 | 10,501 | Complete CDS |
| Serbia reo-like virus 1_Str. 61323 | MT822189 | Culex pipiens | Reoviridae | uncultured virus | 59 | 57 | 4018 | Complete RdRp |
| Serbia reo-like virus 2_Str. 154147 | MT822190 | Cimex lectularius | Reoviridae | Parry’s Lagoon virus | 98 | 49 | 3922 | Complete RdRp |
| Serbia picorna-like virus 2_Str. 153396 | MT822188 | Cimex lectularius | Picornavirales | Hubei picorna-like virus 31 | 73 | 41 | 9931 | Complete CDS |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stanojević, M.; Li, K.; Stamenković, G.; Ilić, B.; Paunović, M.; Pešić, B.; Maslovara, I.Đ.; Šiljić, M.; Ćirković, V.; Zhang, Y. Depicting the RNA Virome of Hematophagous Arthropods from Belgrade, Serbia. Viruses 2020, 12, 975. https://doi.org/10.3390/v12090975
Stanojević M, Li K, Stamenković G, Ilić B, Paunović M, Pešić B, Maslovara IĐ, Šiljić M, Ćirković V, Zhang Y. Depicting the RNA Virome of Hematophagous Arthropods from Belgrade, Serbia. Viruses. 2020; 12(9):975. https://doi.org/10.3390/v12090975
Chicago/Turabian StyleStanojević, Maja, Kun Li, Gorana Stamenković, Bojan Ilić, Milan Paunović, Branislav Pešić, Ivana Đurić Maslovara, Marina Šiljić, Valentina Ćirković, and Yongzhen Zhang. 2020. "Depicting the RNA Virome of Hematophagous Arthropods from Belgrade, Serbia" Viruses 12, no. 9: 975. https://doi.org/10.3390/v12090975
APA StyleStanojević, M., Li, K., Stamenković, G., Ilić, B., Paunović, M., Pešić, B., Maslovara, I. Đ., Šiljić, M., Ćirković, V., & Zhang, Y. (2020). Depicting the RNA Virome of Hematophagous Arthropods from Belgrade, Serbia. Viruses, 12(9), 975. https://doi.org/10.3390/v12090975