Detection of a Reassortant H9N2 Avian Influenza Virus with Intercontinental Gene Segments in a Resident Australian Chestnut Teal

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Ethical Statement
2.2. Virus Enrichment, Nucleic Acid Extraction, cDNA Synthesis, and Non-Targeted Amplification of Pooled and Individual Samples
2.3. cDNA Synthesis and Targeted Avian Influenza Virus PCR Amplification from Individual Samples
2.4. Library Preparation, Next Generation Sequencing (NGS), and Detection of Avian Influenza Virus Sequences
2.5. Alignment of Avian Influenza Virus Reads and Phylogenetic Analysis
2.6. Attempt at Virus Isolation
3. Results
3.1. Detection of Avian Influenza Virus Sequences by Next-Generation Sequencing, Using the Non-Targeted Method
3.2. Detection of Avian Influenza Virus Sequences by Next-Generation Sequencing Using the Targeted Avian Influenza Virus PCR Amplification Method
3.3. Phylogenetic Analysis and Comparison of Avian Influenza Virus Gene Segments
3.4. Identification of Avian Influenza Virus Gene Segment Lineages
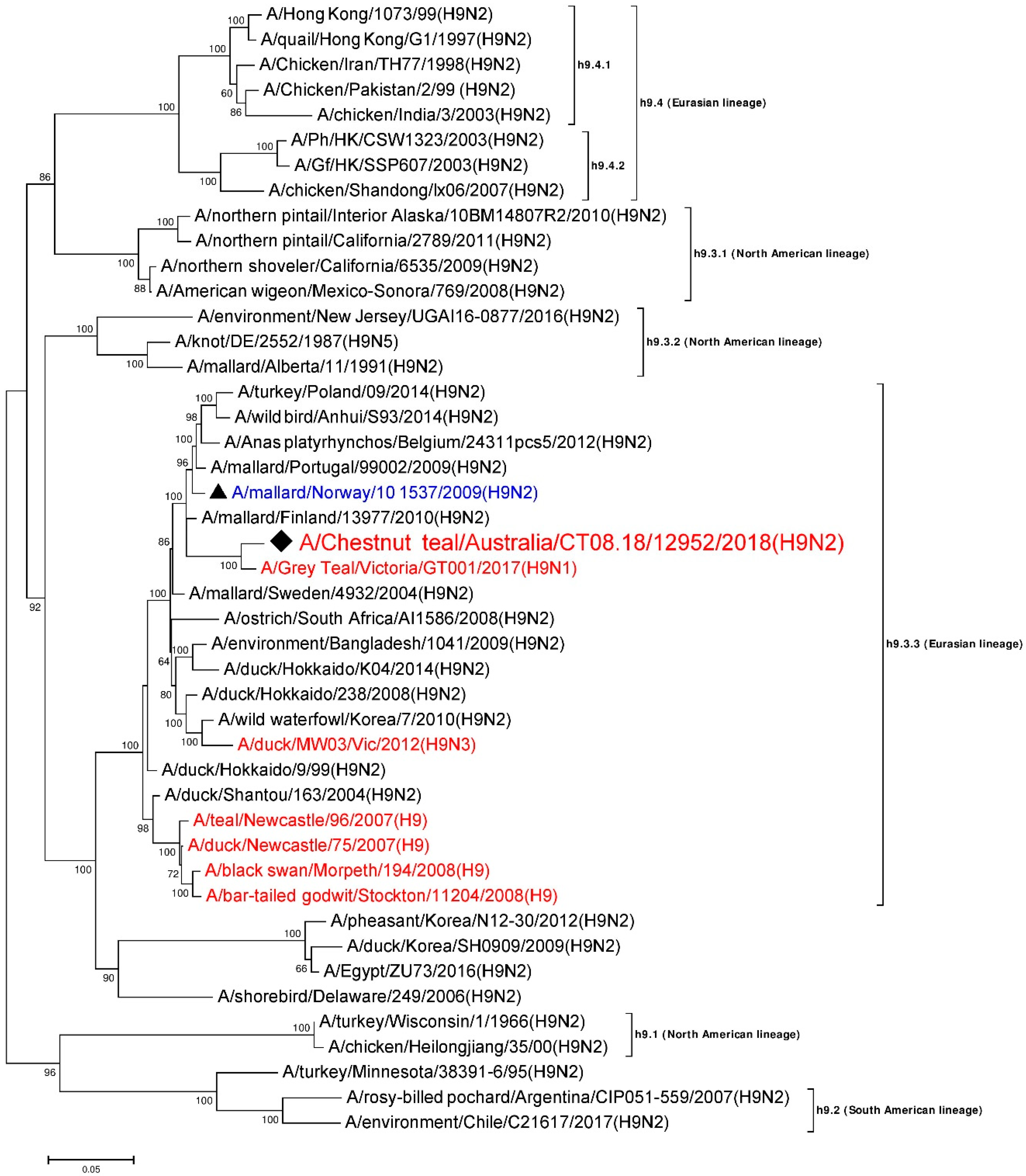
3.5. Phylogenetic Analysis of the H9 Gene Segment
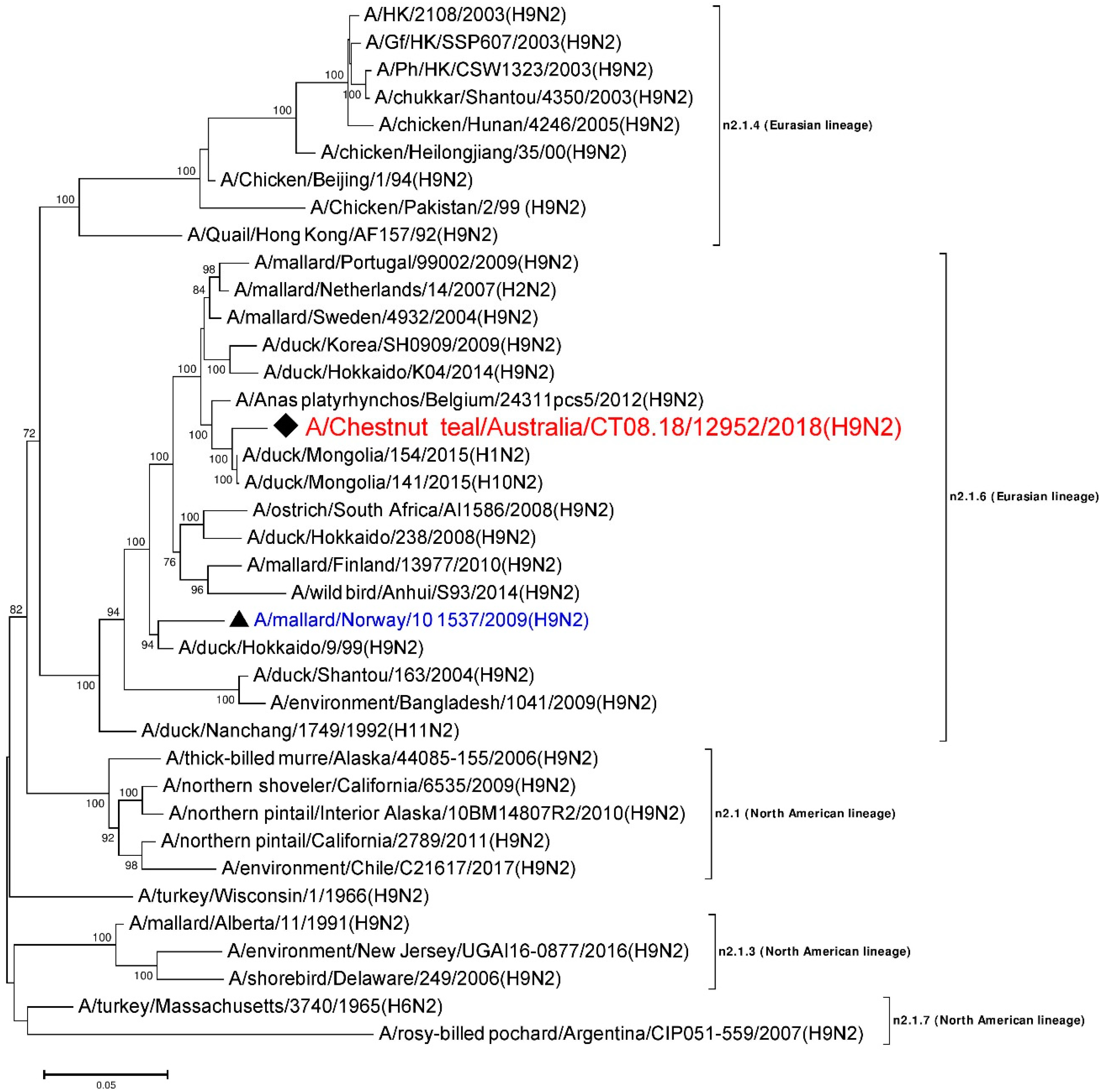
3.6. Phylogenetic Analysis of the N2 Gene
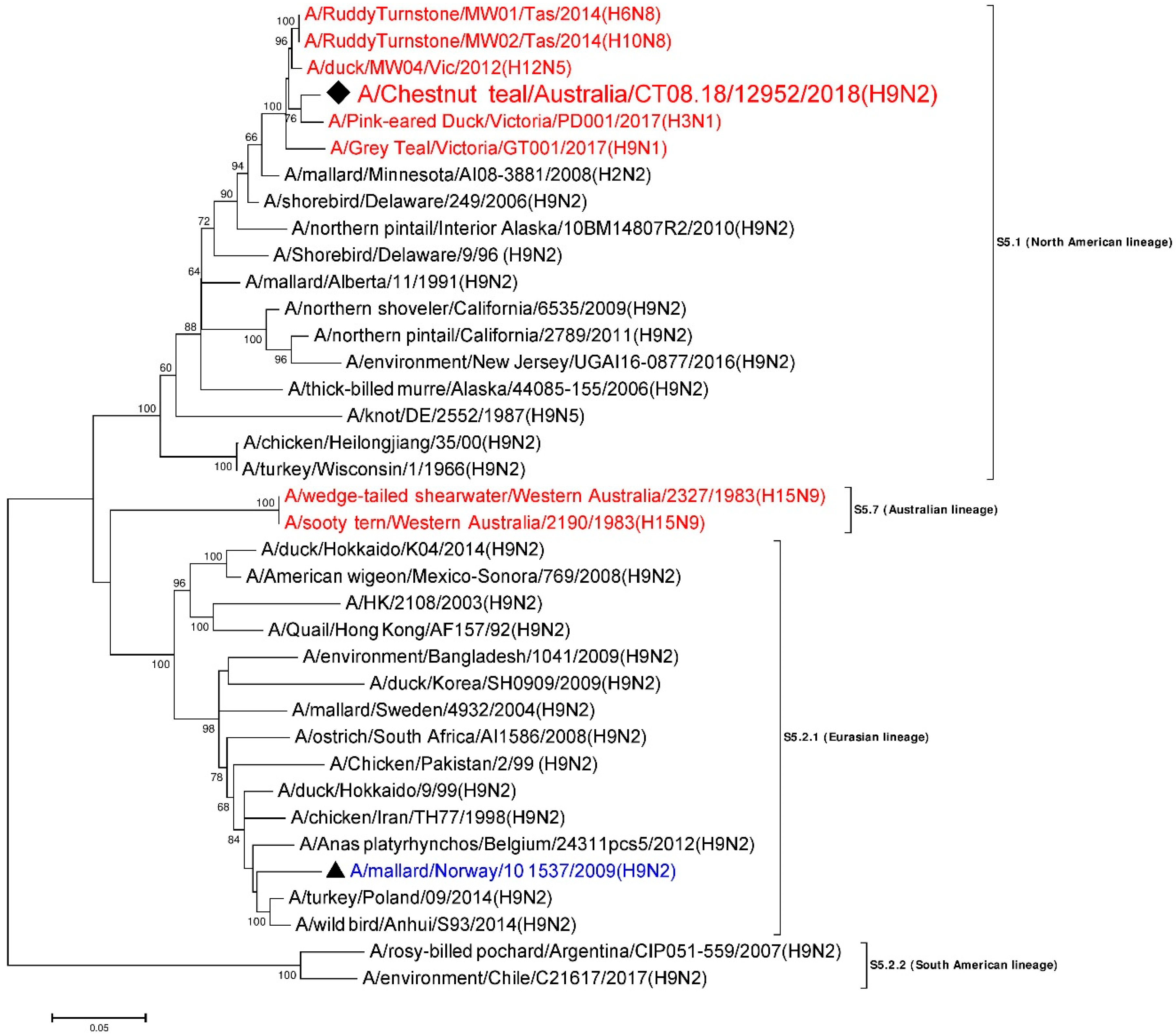
3.7. Phylogenetic Analysis of the Nucleoprotein Gene
3.8. Phylogenetic Analysis and Molecular Characterization of the Remaining 5 Internal Gene Segments of the H9N2 AIV
3.9. Attempt at Virus Isolation
4. Discussion
5. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Tong, S.; Zhu, X.; Li, Y.; Shi, M.; Zhang, J.; Bourgeois, M.; Yang, H.; Chen, X.; Recuenco, S.; Gomez, J. New world bats harbor diverse influenza a viruses. PLoS Pathog. 2013, 9, e1003657. [Google Scholar] [CrossRef] [PubMed]
- Bouvier, N.M.; Palese, P. The biology of influenza viruses. Vaccine 2008, 26, D49–D53. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.I.; Holmes, E.C. The evolution of epidemic influenza. Nat. Rev. Genet. 2007, 8, 196. [Google Scholar] [CrossRef] [PubMed]
- Turner, A. The role of wild aquatic birds in the epidemiology of avian influenza in Australia. Aust. Vet. J. 2004, 82, 713. [Google Scholar] [CrossRef] [PubMed]
- Horimoto, T.; Rivera, E.; Pearson, J.; Senne, D.; Krauss, S.; Kawaoka, Y.; Webster, R.G. Origin and molecular changes associated with emergence of a highly pathogenic h5n2 influenza virus in mexico. Virology 1995, 213, 223–230. [Google Scholar] [CrossRef]
- Garcia, M.; Crawford, J.M.; Latimer, J.W.; RiveraCruz, E.; Perdue, M.L. Heterogeneity in the haemagglutinin gene and emergence of the highly pathogenic phenotype among recent h5n2 avian influenza viruses from mexico. J. Gen. Virol. 1996, 77, 1493–1504. [Google Scholar] [CrossRef]
- Spackman, E.; Senne, D.A.; Davison, S.; Suarez, D.L. Sequence analysis of recent h7 avian influenza viruses associated with three different outbreaks in commercial poultry in the united states. J. Virol. 2003, 77, 13399–13402. [Google Scholar] [CrossRef]
- Banks, J.; Speidel, E.S.; Moore, E.; Plowright, L.; Piccirillo, A.; Capua, I.; Cordioli, P.; Fioretti, A.; Alexander, D.J. Changes in the haemagglutinin and the neuraminidase genes prior to the emergence of highly pathogenic h7n1 avian influenza viruses in italy. Arch. Virol. 2001, 146, 963–973. [Google Scholar] [CrossRef]
- Monne, I.; Fusaro, A.; Nelson, M.I.; Bonfanti, L.; Mulatti, P.; Hughes, J.; Murcia, P.R.; Schivo, A.; Valastro, V.; Moreno, A.; et al. Emergence of a highly pathogenic avian influenza virus from a low-pathogenic progenitor. J. Virol. 2014, 88, 4375–4388. [Google Scholar] [CrossRef]
- Dietze, K.; Graaf, A.; Homeier-Bachmann, T.; Grund, C.; Forth, L.; Pohlmann, A.; Jeske, C.; Wintermann, M.; Beer, M.; Conraths, F.J. From low to high pathogenicity—Characterization of h7n7 avian influenza viruses in two epidemiologically linked outbreaks. Transbound. Emerg. Dis. 2018, 65, 1576–1587. [Google Scholar] [CrossRef]
- Luczo, J.M.; Tachedjian, M.; Harper, J.A.; Payne, J.S.; Butler, J.M.; Sapats, S.I.; Lowther, S.L.; Michalski, W.P.; Stambas, J.; Bingham, J. Evolution of high pathogenicity of h5 avian influenza virus: Haemagglutinin cleavage site selection of reverse-genetics mutants during passage in chickens. Sci. Rep. 2018, 8, 11518. [Google Scholar] [CrossRef]
- Alexander, D.J. An overview of the epidemiology of avian influenza. Vaccine 2007, 25, 5637–5644. [Google Scholar] [CrossRef] [PubMed]
- James, J.; Sealy, J.E.; Iqbal, M. A global perspective on h9n2 avian influenza virus. Viruses 2019, 11, 620. [Google Scholar]
- Guo, Y.; Krauss, S.; Senne, D.; Mo, I.; Lo, K.; Xiong, X.; Norwood, M.; Shortridge, K.; Webster, R.; Guan, Y. Characterization of the pathogenicity of members of the newly established h9n2 influenza virus lineages in asia. Virology 2000, 267, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, X.; Zhong, S.; Sun, H.; Pan, J.; Chen, S.; Peng, D.; Liu, X. Genome sequencing and phylogenetic analysis of avian influenza viruses subtype h9n2. Bing Du Xue Bao Chin. J. Virol. 2012, 28, 7–14. [Google Scholar]
- Lin, Y.; Shaw, M.; Gregory, V.; Cameron, K.; Lim, W.; Klimov, A.; Subbarao, K.; Guan, Y.; Krauss, S.; Shortridge, K. Avian-to-human transmission of h9n2 subtype influenza a viruses: Relationship between h9n2 and h5n1 human isolates. Proc. Natl. Acad. Sci. USA 2000, 97, 9654–9658. [Google Scholar] [CrossRef]
- Butt, K.; Smith, G.J.; Chen, H.; Zhang, L.; Leung, Y.C.; Xu, K.; Lim, W.; Webster, R.G.; Yuen, K.; Peiris, J.M. Human infection with an avian h9n2 influenza a virus in hong kong in 2003. J. Clin. Microbiol. 2005, 43, 5760–5767. [Google Scholar] [CrossRef]
- Potdar, V.; Hinge, D.; Satav, A.; Simões, E.F.; Yadav, P.D.; Chadha, M.S. Laboratory-confirmed avian influenza a (H9N2) virus infection, India, 2019. Emerging Infect. Dis. 2019, 25, 2328. [Google Scholar] [CrossRef]
- Gu, M.; Chen, H.; Li, Q.; Huang, J.; Zhao, M.; Gu, X.; Jiang, K.; Wang, X.; Peng, D.; Liu, X. Enzootic genotype s of h9n2 avian influenza viruses donates internal genes to emerging zoonotic influenza viruses in China. Vet. Microbiol. 2014, 174, 309–315. [Google Scholar] [CrossRef]
- Zhao, G.; Gu, X.; Lu, X.; Pan, J.; Duan, Z.; Zhao, K.; Gu, M.; Liu, Q.; He, L.; Chen, J. Novel reassortant highly pathogenic h5n2 avian influenza viruses in poultry in china. PLoS ONE 2012, 7, e46183. [Google Scholar] [CrossRef]
- Wu, A.; Su, C.; Wang, D.; Peng, Y.; Liu, M.; Hua, S.; Li, T.; Gao, G.F.; Tang, H.; Chen, J. Sequential reassortments underlie diverse influenza h7n9 genotypes in china. Cell Host Microbe 2013, 14, 446–452. [Google Scholar] [CrossRef] [PubMed]
- Sediri, H.; Thiele, S.; Schwalm, F.; Gabriel, G.; Klenk, H.-D. Pb2 subunit of avian influenza virus subtype h9n2: A pandemic risk factor. J. Gen. Virol. 2016, 97, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Yuan, R.; Liang, L.; Wu, J.; Kang, Y.; Song, Y.; Zou, L.; Zhang, X.; Ni, H.; Ke, C. Human infection with an avian influenza a/h9n2 virus in guangdong in 2016. J. Infect. 2017, 74, 422–425. [Google Scholar] [CrossRef] [PubMed]
- Shanmuganatham, K.K.; Jones, J.C.; Marathe, B.M.; Feeroz, M.M.; Jones-Engel, L.; Walker, D.; Turner, J.; Rabiul Alam, S.; Kamrul Hasan, M.; Akhtar, S. The replication of bangladeshi h9n2 avian influenza viruses carrying genes from h7n3 in mammals. Emerg. Microbes Infect. 2016, 5, 1–12. [Google Scholar] [CrossRef]
- Tracey, J.P.; Woods, R.; Roshier, D.; West, P.; Saunders, G.R. The role of wild birds in the transmission of avian influenza for australia: An ecological perspective. Emu 2004, 104, 109–124. [Google Scholar] [CrossRef]
- Yoon, S.-W.; Webby, R.J.; Webster, R.G. Evolution and ecology of influenza a viruses. In Influenza Pathogenesis and Control-Volume I; Springer: Berlin/Heidelberg, Germany, 2014; pp. 359–375. [Google Scholar]
- Wille, M.; Eden, J.S.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virus–virus interactions and host ecology are associated with rna virome structure in wild birds. Mol. Ecol. 2018, 27, 5263–5278. [Google Scholar] [CrossRef]
- Wille, M.; Shi, M.; Klaassen, M.; Hurt, A.C.; Holmes, E.C. Virome heterogeneity and connectivity in waterfowl and shorebird communities. ISME J. 2019, 13, 2603–2616. [Google Scholar] [CrossRef]
- Wille, M.; Lisovski, S.; Risely, A.; Ferenczi, M.; Roshier, D.; Wong, F.Y.; Breed, A.C.; Klaassen, M.; Hurt, A.C. Serologic evidence of exposure to highly pathogenic avian influenza h5 viruses in migratory shorebirds, australia. Emerg. Infect. Dis. 2019, 25, 1903. [Google Scholar] [CrossRef]
- Norman, F.; Brown, R. Aspects of the distribution and abundance of chestnut teal in south-eastern australia. Emu-Austral Ornithol. 1988, 88, 70–80. [Google Scholar] [CrossRef]
- Hansen, B.D.; Menkhorst, P.; Moloney, P.; Loyn, R.H. Long-term declines in multiple waterbird species in a tidal embayment, south-east a ustralia. Austral. Ecol. 2015, 40, 515–527. [Google Scholar] [CrossRef]
- Beaumont, L.J.; McALLAN, I.A.; Hughes, L. A matter of timing: Changes in the first date of arrival and last date of departure of australian migratory birds. Glob. Chang. Biol. 2006, 12, 1339–1354. [Google Scholar] [CrossRef]
- Liu, S.; Ji, K.; Chen, J.; Tai, D.; Jiang, W.; Hou, G.; Chen, J.; Li, J.; Huang, B. Panorama phylogenetic diversity and distribution of type a influenza virus. PLoS ONE 2009, 4, e5022. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.M.; Sun, Y.X.; Chen, J.W.; Liu, S.; Yu, J.M.; Shen, C.J.; Sun, X.D.; Peng, D. Panorama phylogenetic diversity and distribution of type a influenza viruses based on their six internal gene sequences. Virol. J. 2009, 6, 137. [Google Scholar] [CrossRef] [PubMed]
- Hansbro, P.M.; Warner, S.; Tracey, J.P.; Arzey, K.E.; Selleck, P.; O’Riley, K.; Beckett, E.L.; Bunn, C.; Kirkland, P.D.; Vijaykrishna, D.; et al. Surveillance and analysis of avian influenza viruses, australia. Emerg Infect Dis. 2010, 16, 1896–1904. [Google Scholar] [CrossRef] [PubMed]
- Bhatta, T.R.; Chamings, A.; Vibin, J.; Alexandersen, S. Detection and characterisation of canine astrovirus, canine parvovirus and canine papillomavirus in puppies using next generation sequencing. Sci. Rep. 2019, 9, 4602. [Google Scholar] [CrossRef] [PubMed]
- Vibin, J.; Chamings, A.; Collier, F.; Klaassen, M.; Nelson, T.M.; Alexandersen, S. Metagenomics detection and characterisation of viruses in faecal samples from australian wild birds. Sci. Rep. 2018, 8, 8686. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Mount, D.W. Using the basic local alignment search tool (blast). Cold Spring Harb. Protoc. 2007, 2007. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. Mega7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Caboche, S.; Audebert, C.; Lemoine, Y.; Hot, D. Comparison of mapping algorithms used in high-throughput sequencing: Application to ion torrent data. BMC Genom. 2014, 15, 264. [Google Scholar] [CrossRef]
- Thorvaldsdóttir, H.; Robinson, J.T.; Mesirov, J.P. Integrative genomics viewer (igv): High-performance genomics data visualization and exploration. Brief. Bioinform. 2013, 14, 178–192. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R. Clustal w and clustal x version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Xu, K.M.; Ferreri, L.; Rimondi, A.; Olivera, V.; Romano, M.; Ferreyra, H.; Rago, V.; Uhart, M.; Chen, H.J.; Sutton, T.; et al. Isolation and characterization of an h9n2 influenza virus isolated in argentina. Virus Res. 2012, 168, 41–47. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar] [PubMed]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and g+c-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar] [PubMed]
- Gonzalez-Reiche, A.S.; Perez, D.R. Where do avian influenza viruses meet in the americas? Avian Dis. 2012, 56, 1025–1033. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: Oxford, UK, 2000. [Google Scholar]
- Eisfeld, A.J.; Neumann, G.; Kawaoka, Y. Influenza a virus isolation, culture and identification. Nat. Protoc. 2014, 9, 2663. [Google Scholar] [CrossRef]
- Lindh, E.; Ek-Kommonen, C.; Vaananen, V.M.; Vaheri, A.; Vapalahti, O.; Huovilainen, A. Molecular epidemiology of h9n2 influenza viruses in northern europe. Vet. Microbiol. 2014, 172, 548–554. [Google Scholar] [CrossRef]
- Van Borm, S.; Rosseel, T.; Marché, S.; Steensels, M.; Vangeluwe, D.; Linden, A.; van den Berg, T.; Lambrecht, B. Complete coding sequences of one h9 and three h7 low-pathogenic influenza viruses circulating in wild birds in belgium, 2009 to 2012. Genome Announc. 2016, 4, e00540-16. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide-sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Gene Segment | Genbank Virus Sequence with Highest Nucleotide Identity | Accession Number | Sub-Lineages * | Nucleotide Match | Identity % |
|---|---|---|---|---|---|
| PB2 | A/duck/Bangladesh/26992/2015 (H7N9) | KY635525 | S1.2.4 (Eurasian) | 2280/2308 | 98.79 |
| HA | A/Grey Teal/Victoria/GT001/2017 (H9N1) | MK213322 | h9.3.3 (Eurasian) | 1667/1703 | 97.89 |
| NP | A/Ruddy Turnstone/MW02/Tas/2014 (H10N8) | MH453824 | S5.1 (North American) | 1517/1546 | 98.12 |
| NA | A/duck/Mongolia/154/2015 (H1N2) | LC121278 | n2.1.6 (Eurasian) | 1441/1463 | 98.49 |
| M1–M2 | A/Grey Teal/Victoria/GT001/2017 (H9N1) | MK213323 | S7.2.5 (Eurasian) | 969/982 | 98.68 |
| NEP-NS1 | A/Ruddy Turnstone/MW02/Tas/2014 (H10N8) | MH453825 | S8.2.1 (Eurasian) | 872/882 | 98.87 |
| Gene Segment | Genbank Virus Sequence with Highest Nucleotide Identity | Accession Number | Sub-Lineages * | Nucleotide Match | Identity % |
|---|---|---|---|---|---|
| PB1 | A/duck/Mongolia/154/2015 (H1N2) | LC121274 | S2.2.2 (Eurasian) | 1338/1381 | 96.88 |
| PA | A/Grey Teal/Victoria/GT001/2017 (H9N1) | MK213327 | S3.2.7 (Eurasian) | 1120/1130 | 99.12 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhatta, T.R.; Chamings, A.; Vibin, J.; Klaassen, M.; Alexandersen, S. Detection of a Reassortant H9N2 Avian Influenza Virus with Intercontinental Gene Segments in a Resident Australian Chestnut Teal. Viruses 2020, 12, 88. https://doi.org/10.3390/v12010088
Bhatta TR, Chamings A, Vibin J, Klaassen M, Alexandersen S. Detection of a Reassortant H9N2 Avian Influenza Virus with Intercontinental Gene Segments in a Resident Australian Chestnut Teal. Viruses. 2020; 12(1):88. https://doi.org/10.3390/v12010088
Chicago/Turabian StyleBhatta, Tarka Raj, Anthony Chamings, Jessy Vibin, Marcel Klaassen, and Soren Alexandersen. 2020. "Detection of a Reassortant H9N2 Avian Influenza Virus with Intercontinental Gene Segments in a Resident Australian Chestnut Teal" Viruses 12, no. 1: 88. https://doi.org/10.3390/v12010088
APA StyleBhatta, T. R., Chamings, A., Vibin, J., Klaassen, M., & Alexandersen, S. (2020). Detection of a Reassortant H9N2 Avian Influenza Virus with Intercontinental Gene Segments in a Resident Australian Chestnut Teal. Viruses, 12(1), 88. https://doi.org/10.3390/v12010088