Recombination Events and Conserved Nature of Receptor Binding Motifs in Coxsackievirus A9 Isolates



Abstract
1. Introduction
2. Materials and Methods
2.1. Clinical Specimens and Symptoms
2.2. Amplification of VP1 and 3Dpol Regions
2.3. Nucleotide Sequencing
2.4. Phylogenetic Analysis
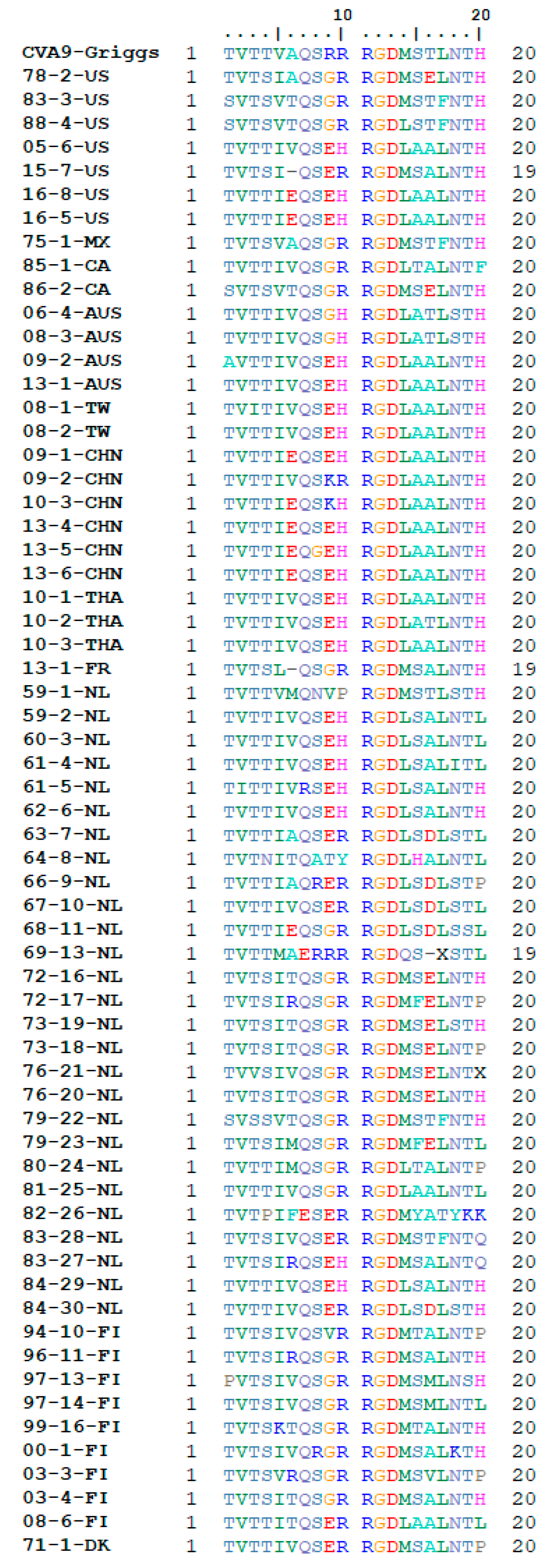
2.5. RGD and HS Binding Site Analysis
3. Results
3.1. Phylogeny of CVA9 VP1 and 3Dpol Regions
3.2. Receptor Binding Site Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Pallansch, M.A. Molecular evolution of the human enteroviruses: Correlation of serotype with VP1 sequence and application to picornavirus classification. J. Virol. 1999, 73, 1941–1948. [Google Scholar] [CrossRef]
- Oberste, M.S.; Nix, W.A.; Maher, K.; Pallansch, M.A. Improved molecular identification of enteroviruses by RT-PCR and amplicon sequencing. J. Clin. Virol. 2003, 26, 375–377. [Google Scholar] [CrossRef]
- Palacios, G.; Oberste, M. Enteroviruses as agents of emerging infectious diseases. J. Neurovirol. 2005, 11, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Tuthill, T.J.; Groppelli, E.; Hogle, J.M.; Rowlands, D.J. Picornaviruses. In Cell Entry by Non-Enveloped Viruses. Current Topics in Microbiology and Immunology, vol 343; Johnson, J., Ed.; Springer: Berlin/Heidelberg, Germany, 2010; pp. 43–89. [Google Scholar]
- Chang, K.H.; Auvinen, P.; Hyypia, T.; Stanway, G. The nucleotide sequence of Coxsackievirus A9; Implications for receptor binding and enterovirus classification. J. Gen. Virol. 1989, 70, 3269–3280. [Google Scholar] [CrossRef]
- Pulli, T.; Koskimies, P.; Hyypiä, T. Molecular Comparison of Coxsackie A Virus Serotypes. Virology 1995, 212, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Blomqvist, S.; Paananen, A.; Savolainen-Kopra, C.; Hovi, T.; Roivainen, M. Eight Years of Experience with Molecular Identification of Human Enteroviruses. J. Clin. Microbiol. 2008, 46, 2410–2413. [Google Scholar] [CrossRef] [PubMed]
- Cui, A.; Yu, D.; Zhu, Z.; Meng, L.; Li, H.; Liu, J.; Liu, G.; Mao, N.; Xu, W. An outbreak of aseptic meningitis caused by coxsackievirus A9 in Gansu, the People’s Republic of China. Virol. J. 2010, 7, 72. [Google Scholar] [CrossRef]
- Aoki, Y.; Abe, A.; Ikeda, T.; Abiko, C.; Mizuta, K.; Yamaguchi, I.; Ahiko, T. An Outbreak of Exanthematous Disease due to Coxsackievirus A9 in a Nursery in Yamagata, Japan, from February to March 2012. Jpn. J. Infect. Dis. 2012, 65, 367–369. [Google Scholar] [CrossRef]
- Merilahti, P.; Koskinen, S.; Heikkilä, O.; Karelehto, E.; Susi, P. Endocytosis of integrin-binding human picornaviruses. Adv. Virol. 2012, 2012. [Google Scholar] [CrossRef]
- Hendry, E.; Hatanaka, H.; Fry, E.; Smyth, M.; Tate, J.; Stanway, G.; Santti, J.; Maaronen, M.; Hyypiä, T.; Stuart, D. The crystal structure of coxsackievirus A9: New insights into the uncoating mechanisms of enteroviruses. Structure 1999, 7, 1527–1538. [Google Scholar] [CrossRef][Green Version]
- Shakeel, S.; Seitsonen, J.J.T.; Kajander, T.; Laurinmaki, P.; Hyypia, T.; Susi, P.; Butcher, S.J. Structural and Functional Analysis of Coxsackievirus A9 Integrin v 6 Binding and Uncoating. J. Virol. 2013, 87, 3943–3951. [Google Scholar] [CrossRef] [PubMed]
- Heikkilä, O.; Susi, P.; Stanway, G.; Hyypiä, T. Integrin αVβ6 is a high-affinity receptor for coxsackievirus A9. J. Gen. Virol. 2009, 90, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Heikkilä, O.; Susi, P.; Tevaluoto, T.; Härmä, H.; Marjomäki, V.; Hyypiä, T.; Kiljunen, S. Internalization of coxsackievirus A9 is mediated by β2-microglobulin, dynamin, and Arf6 but not by caveolin-1 or clathrin. J. Virol. 2010, 84, 3666–3681. [Google Scholar] [CrossRef] [PubMed]
- Roivainen, M.; Hyypiä, T.; Piirainen, L.; Kalkkinen, N.; Stanway, G.; Hovi, T. RGD-dependent entry of coxsackievirus A9 into host cells and its bypass after cleavage of VP1 protein by intestinal proteases. J. Virol. 1991, 65, 4735–4740. [Google Scholar] [PubMed]
- Roivainen, M.; Piirainen, L.; Hovi, T.; Virtanen, I.; Riikonen, T.; Heino, J.; Hyypiä, T. Entry of coxsackievirus A9 into host cells: Specific interactions with αVβ3 integrin, the vitronectin receptor. Virology 1994, 203, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Hughes, P.J.; Horsnell, C.; Hyypiä, T.; Stanway, G. The coxsackievirus A9 RGD motif is not essential for virus viability. J. Virol. 1995, 69, 8035–8040. [Google Scholar]
- Heikkilä, O.; Merilahti, P.; Hakanen, M.; Karelehto, E.; Alanko, J.; Sukki, M.; Kiljunen, S.; Susi, P. Integrins are not essential for entry of coxsackievirus A9 into SW480 human colon adenocarcinoma cells. Virol. J. 2016, 13, 171. [Google Scholar] [CrossRef]
- Triantafilou, K.; Fradelizi, D.; Wilson, K.; Triantafilou, M. GRP78, a Coreceptor for Coxsackievirus A9, Interacts with Major Histocompatibility Complex Class I Molecules Which Mediate Virus Internalization. J. Virol. 2002, 76, 633–643. [Google Scholar] [CrossRef]
- McLeish, N.J.; Williams, C.H.; Kaloudas, D.; Roivainen, M.M.; Stanway, G. Symmetry-related clustering of positive charges is a common mechanism for heparan sulfate binding in enteroviruses. J. Virol. 2012, 86, 11163–11170. [Google Scholar] [CrossRef][Green Version]
- Merilahti, P.; Karelehto, E.; Susi, P. Role of Heparan Sulfate in Cellular Infection of Integrin-Binding Coxsackievirus A9 and Human Parechovirus 1 Isolates. PLoS ONE 2016, 11. [Google Scholar] [CrossRef]
- Santti, J.; Harvala, H.; Kinnunen, L.; Hyypiä, T. Molecular epidemiology and evolution of coxsackievirus A9. J. Gen. Virol. 2000, 81, 1361–1372. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. Rates of spontaneous mutation among RNA viruses. Proc. Natl. Acad. Sci. USA 1993, 90, 4171–4175. [Google Scholar] [CrossRef] [PubMed]
- Zoll, J.; Galama, J.; Melchers, W. Intratypic genome variability of the coxsackievirus B1 2A protease region. J. Gen. Virol. 1994, 75, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Hughes, M.S.; Hoey, E.M.; Coyle, P.V. A nucleotide sequence comparison of coxsackievirus B4 isolates from aquatic samples and clinical specimens. Epidemiol. Infect. 1993, 110, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Kopecka, H.; Brown, B.; Pallansch, M. Genotypic variation in Coxsackievirus B5 isolates from three different outbreaks in the United States. Virus Res. 1995, 38, 125–136. [Google Scholar] [CrossRef]
- Brown, B.A.; Oberste, M.S.; Alexander, J.P.; Kennett, M.L.; Pallansch, M.A. Molecular epidemiology and evolution of enterovirus 71 strains isolated from 1970 to 1998. J. Virol. 1999, 73, 9969–9975. [Google Scholar] [CrossRef]
- Gjøen, K.; Bruu, A.-L.; Ørstavik, I. Intratypic genome variability of echovirus type 30 in part of the VP4/VP2 coding region. Arch. Virol. 1996, 141, 901–908. [Google Scholar] [CrossRef]
- Santti, J.; Hyypiä, T.; Kinnunen, L.; Salminen, M. Evidence of recombination among enteroviruses. J. Virol. 1999, 73, 8741–8749. [Google Scholar] [CrossRef]
- Fernandez, N.; Triantafilou, K.; Stanway, G.; Wilson, K.M.; Triantafilou, M.; Takada, Y. Involvement of β2-microglobulin and integrin αvβ3 molecules in the coxsackievirus A9 infectious cycle. J. Gen. Virol. 1999, 80, 2591–2600. [Google Scholar]
- Triantafilou, M.; Triantafilou, K.; Wilson, K.M.; Takada, Y.; Fernandez, N. High affinity interactions of Coxsackievirus A9 with integrin αvβ3 (CD51/61) require the CYDMKTTC sequence of β3, but do not require the RGD sequence of the CAV-9 VP1 protein. Hum. Immunol. 2000, 61, 453–459. [Google Scholar] [CrossRef]
- Triantafilou, M.; Wilson, K.M.; Triantafilou, K. Identification of echovirus 1 and coxsackievirus A9 receptor molecules via a novel flow cytometric quantification method. Cytometry 2001, 43, 279–289. [Google Scholar] [CrossRef]
- Harvala, H. Pathogenesis of coxsackievirus A9 in mice: Role of the viral arginine-glycine-aspartic acid motif. J. Gen. Virol. 2003, 84, 2375–2379. [Google Scholar] [CrossRef] [PubMed]
- Harvala, H. Tissue tropism of recombinant coxsackieviruses in an adult mouse model. J. Gen. Virol. 2005, 86, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Leitch, E.C.M.; Harvala, H.; Robertson, I.; Ubillos, I.; Templeton, K.; Simmonds, P. Erratum to “Direct identification of human enterovirus serotypes in cerebrospinal fluid by amplification and sequencing of the VP1 region” [J. Clin. Virol. 44 (2009) 119–124]. J. Clin. Virol. 2011, 51, 286. [Google Scholar] [CrossRef]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView Version 4: A Multiplatform Graphical User Interface for Sequence Alignment and Phylogenetic Tree Building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef]
- Shapiro, B.; Rambaut, A.; Pybus, O.G.; Holmes, E.C. A phylogenetic method for detecting positive epistasis in gene sequences and its application to RNA virus evolution. Mol. Biol. Evol. 2006, 23, 1724–1730. [Google Scholar] [CrossRef]
- Okonechnikov, K.; Golosova, O.; Fursov, M.; Varlamov, A.; Vaskin, Y.; Efremov, I.; German Grehov, O.G.; Kandrov, D.; Rasputin, K.; Syabro, M.; et al. Unipro UGENE: A unified bioinformatics toolkit. Bioinformatics 2012, 28, 1166–1167. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Williams, C.H.; Kajander, T.; Hyypia, T.; Jackson, T.; Sheppard, D.; Stanway, G. Integrin v 6 is an RGD-Dependent Receptor for Coxsackievirus A9. J. Virol. 2004, 78, 6967–6973. [Google Scholar] [CrossRef] [PubMed]
- Pybus, O.G.; Tatem, A.J.; Lemey, P. Virus evolution and transmission in an ever more connected world. Proc. R. Soc. B Biol. Sci. 2015, 282. [Google Scholar] [CrossRef] [PubMed]
- Jackson, T.; Blakemore, W.; Newman, J.W.I.; Knowles, N.J.; Mould, A.P.; Humphries, M.J.; King, A.M.Q. Foot-and-mouth disease virus is a ligand for the high-affinity binding conformation of integrin αVβ1: Influence of the leucine residue within the RGDL motif on selectivity of integrin binding. J. Gen. Virol. 2000, 81, 1383–1391. [Google Scholar] [CrossRef] [PubMed]
- DiCara, D.; Burman, A.; Clark, S.; Berryman, S.; Howard, M.J.; Hart, I.R.; Marshall, J.F.; Jackson, T. Foot-and-mouth disease virus forms a highly stable, EDTA-resistant complex with its principal receptor, integrin αVβ6: Implications for infectiousness. J. Virol. 2008, 82, 1537–1546. [Google Scholar] [CrossRef]
- Cardin, A.D.; Weintraub, H.J. Molecular modeling of protein-glycosaminoglycan interactions. Arteriosclerosis 1989, 9, 21–32. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Sample ID 1 | Isolation Year | Country of Origin | Sample Type | Clinical Symptoms | GenBank Accession 2 |
|---|---|---|---|---|---|
| 06-4-AUS | 2008 | Australia | Feces | N/A | MF678346.1 |
| 08-3-AUS | 2008 | Australia | Feces | N/A | MF678309.1 |
| 09-2-AUS | 2009 | Australia | Feces | N/A | MF678303.1 |
| 13-1-AUS | 2013 | Australia | Feces | N/A | MF678330.1 |
| 85-1-CA | 1985 | Canada | Feces | Meningitis | MN494025/MN494079 |
| 86-2-CA | 1986 | Canada | Feces | Headache, diarrhea, URI | MN494026/MN494080 |
| 09-1-CHN | 2009 | China | Feces | Meningitis | KM890277.1 |
| 09-2-CHN | 2009 | China | Feces | Meningitis | KM890278.1 |
| 10-3-CHN | 2010 | China | N/A | N/A | KP266574.1 |
| 13-4-CHN | 2013 | China | CSF | HFMD | KP289437.1 |
| 13-5-CHN | 2013 | China | CSF | HFMD | KP290111.1 |
| 13-6-CHN | 2013 | China | CSF | HFMD | KP289434.1 |
| 71-1-DK | 1971 | Denmark | Feces | Fever | MN494022/MN494076 |
| 00-1-FI | 2000 | Finland | Feces | Gastroenteritis | MN493979/MN494033 |
| 02-2-FI | 2002 | Finland | Feces | Diarrhea | MN493980/MN494034 |
| 03-3-FI | 2003 | Finland | Feces | Diarrhea | MN493981/MN494035 |
| 03-4-FI | 2003 | Finland | Feces | Diarrhea | MN493982/MN494036 |
| 07-5-FI | 2007 | Finland | Feces | Diarrhea | MN493983/MN494037 |
| 08-6-FI | 2008 | Finland | CSF | Meningitis | MN493984/MN494038 |
| 08-7-FI | 2008 | Finland | CSF | Meningitis | MN493985/MN494039 |
| 08-8-FI | 2008 | Finland | CSF | Meningitis | MN493986/MN494040 |
| 08-9-FI | 2008 | Finland | CSF | Meningitis | MN493987/MN494041 |
| 94-10-FI | 1994 | Finland | Feces | Cerebellitis | MN494030/MN494084 |
| 96-11-FI | 1996 | Finland | Feces | Meningitis | MN494032/MN494086 |
| 97-12-FI | 1997 | Finland | Throat swab | Meningitis | MN494018/MN494072 |
| 97-13-FI | 1997 | Finland | CSF | Meningitis | MN494019/MN494073 |
| 97-14-FI | 1997 | Finland | CSF | Meningitis | MN494031/MN494085 |
| 99-15-FI | 1999 | Finland | Nasal swab | Exanthema | MN494020/MN494074 |
| 99-16-FI | 1999 | Finland | Nasal swab | Exanthema | MN494021/MN494075 |
| 13-1-FR | 2013 | France | N/A | N/A | KM201659.1 |
| 75-1-MX | 1975 | Mexico | Feces | Diarrhea | MN494027/MN494081 |
| 59-1-NL | 1959 | Netherlands | Feces | Diarrhea, exanthema | MN493988/MN494042 |
| 59-2-NL | 1959 | Netherlands | Feces | Exanthema, leucopenia | MN493989/MN494043 |
| 60-3-NL | 1960 | Netherlands | CSF | Meningitis | MN493990/MN494044 |
| 61-4-NL | 1961 | Netherlands | CSF | Meningitis | MN493991/MN494045 |
| 61-5-NL | 1961 | Netherlands | CSF | Meningitis | MN493992/MN494046 |
| 62-6-NL | 1962 | Netherlands | Feces | Fever, vomiting | MN493993/MN494047 |
| 63-7-NL | 1963 | Netherlands | Feces | Fever, vomiting | MN493994/MN494048 |
| 64-8-NL | 1964 | Netherlands | Feces | Fever, vomiting | MN493995/MN494049 |
| 66-9-NL | 1966 | Netherlands | Feces | Gastroenteritis | MN493996/MN494050 |
| 67-10-NL | 1967 | Netherlands | Feces | Myelitis, fever | MN493997/MN494051 |
| 68-11-NL | 1968 | Netherlands | Feces | Diarrhea | MN493998/MN494052 |
| 69-12-NL | 1969 | Netherlands | Throat swab | Convulsion | MN493999/MN494053 |
| 69-13-NL | 1969 | Netherlands | CSF | Headache | MN494000/MN494054 |
| 70-14-NL | 1970 | Netherlands | CSF | Meningitis | MN494001/MN494055 |
| 71-15-NL | 1971 | Netherlands | Feces | Meningitis | MN494002/MN494056 |
| 72-16-NL | 1972 | Netherlands | Feces | Meningitis | MN494003/MN494057 |
| 72-17-NL | 1972 | Netherlands | Feces | Facial paralysis | MN494004/MN494058 |
| 73-18-NL | 1973 | Netherlands | Feces | Meningitis | MN494005/MN494059 |
| 73-19-NL | 1973 | Netherlands | Feces | Meningitis | MN494006/MN494060 |
| 76-20-NL | 1976 | Netherlands | Throat swab | Gastroenteritis | MN494007/MN494061 |
| 76-21-NL | 1976 | Netherlands | Feces | Thrombocytopenia | MN494008/MN494062 |
| 79-22-NL | 1979 | Netherlands | Throat swab | Meningitis | MN494009/MN494063 |
| 79-23-NL | 1979 | Netherlands | Feces | Pneumonia | MN494010/MN494064 |
| 80-24-NL | 1980 | Netherlands | Throat swab | Encephalitis | MN494011/MN494065 |
| 81-25-NL | 1981 | Netherlands | Feces | Respiratory symptoms | MN494012/MN494066 |
| 82-26-NL | 1982 | Netherlands | Feces | Diarrhea | MN494013/MN494067 |
| 83-27-NL | 1983 | Netherlands | Feces | Gastroenteritis | MN494014/MN494068 |
| 83-28-NL | 1983 | Netherlands | Feces | N/A | MN494015/MN494069 |
| 84-29-NL | 1984 | Netherlands | urine | Dyspnoea | MN494016/MN494070 |
| 84-30-NL | 1984 | Netherlands | Feces | N/A | MN494017/MN494071 |
| 08-1-TW | 2008 | Taiwan | N/A | N/A | KT353721.1 |
| 08-2-TW | 2008 | Taiwan | N/A | N/A | MF422557.1 |
| 10-1-THA | 2010 | Thailand | Throat/nasal swab | N/A | KU574636.1 |
| 10-2-THA | 2010 | Thailand | Nasal swab | Influenza-like illness | KU574637.1 |
| 10-3-THA | 2010 | Thailand | Nasal swab | Influenza-like illness | KU574638.1 |
| 05-6-US | 2005 | USA | N/A | N/A | MH752987.1 |
| 15-7-US | 2015 | USA | Feces | N/A | MN166093.1 |
| 16-5-US | 2016 | USA | N/A | N/A | KY674974.1 |
| 16-8-US | 2016 | USA | N/A | N/A | KY674976.1 |
| 74-1-US | 1974 | USA | Feces | Headache | MN494024/MN494078 |
| 78-2-US | 1978 | USA | Feces | Diarrhea | MN494029/MN494083 |
| 83-3-US | 1983 | USA | Feces | Meningitis | MN494023/MN494077 |
| 88-4-US | 1988 | USA | Feces | Headache | MN494028/MN494082 |
| CVA9-Griggs | 1948 | USA | N/A | N/A | D00627.1 |
| tMRCA (year) 1 | Substitution Rate (10−3 subs./site/year) 2 | ||
|---|---|---|---|
| VP1 | 3Dpol | VP1 | 3Dpol |
| 1889 (1856–1918) | 1814 (1726–1886) | 4.1 (3.1–5.0) | 3.4 (2.3–4.6) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hietanen, E.; Susi, P. Recombination Events and Conserved Nature of Receptor Binding Motifs in Coxsackievirus A9 Isolates. Viruses 2020, 12, 68. https://doi.org/10.3390/v12010068
Hietanen E, Susi P. Recombination Events and Conserved Nature of Receptor Binding Motifs in Coxsackievirus A9 Isolates. Viruses. 2020; 12(1):68. https://doi.org/10.3390/v12010068
Chicago/Turabian StyleHietanen, Eero, and Petri Susi. 2020. "Recombination Events and Conserved Nature of Receptor Binding Motifs in Coxsackievirus A9 Isolates" Viruses 12, no. 1: 68. https://doi.org/10.3390/v12010068
APA StyleHietanen, E., & Susi, P. (2020). Recombination Events and Conserved Nature of Receptor Binding Motifs in Coxsackievirus A9 Isolates. Viruses, 12(1), 68. https://doi.org/10.3390/v12010068