Metaviromics Reveals Unknown Viral Diversity in the Biting Midge Culicoides impunctatus

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Method
2.1. Sample Collection and Sequencing
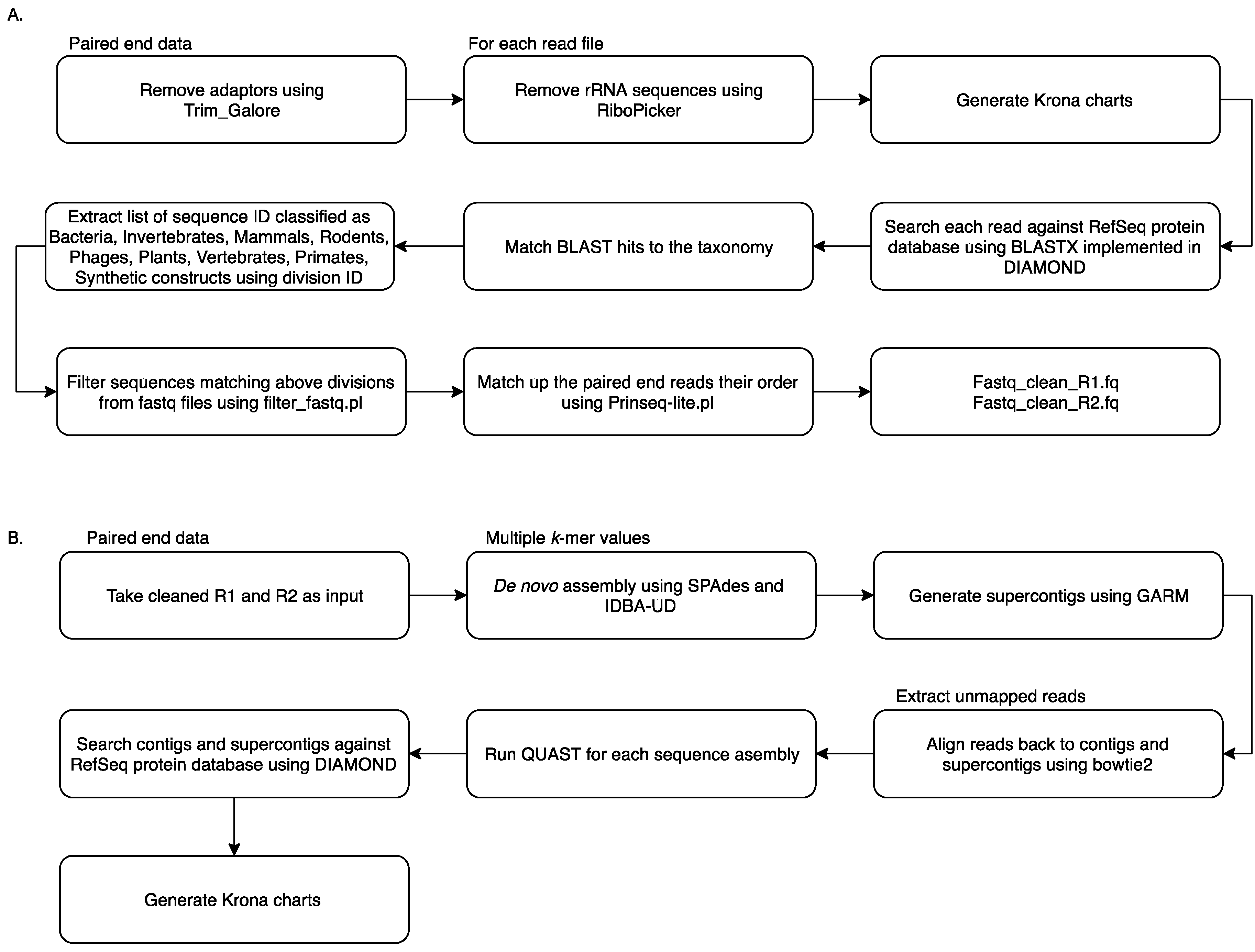
2.2. Metagenomic Analysis Using MetaViC Pipeline
2.3. Protein Domain Identification
3. Results
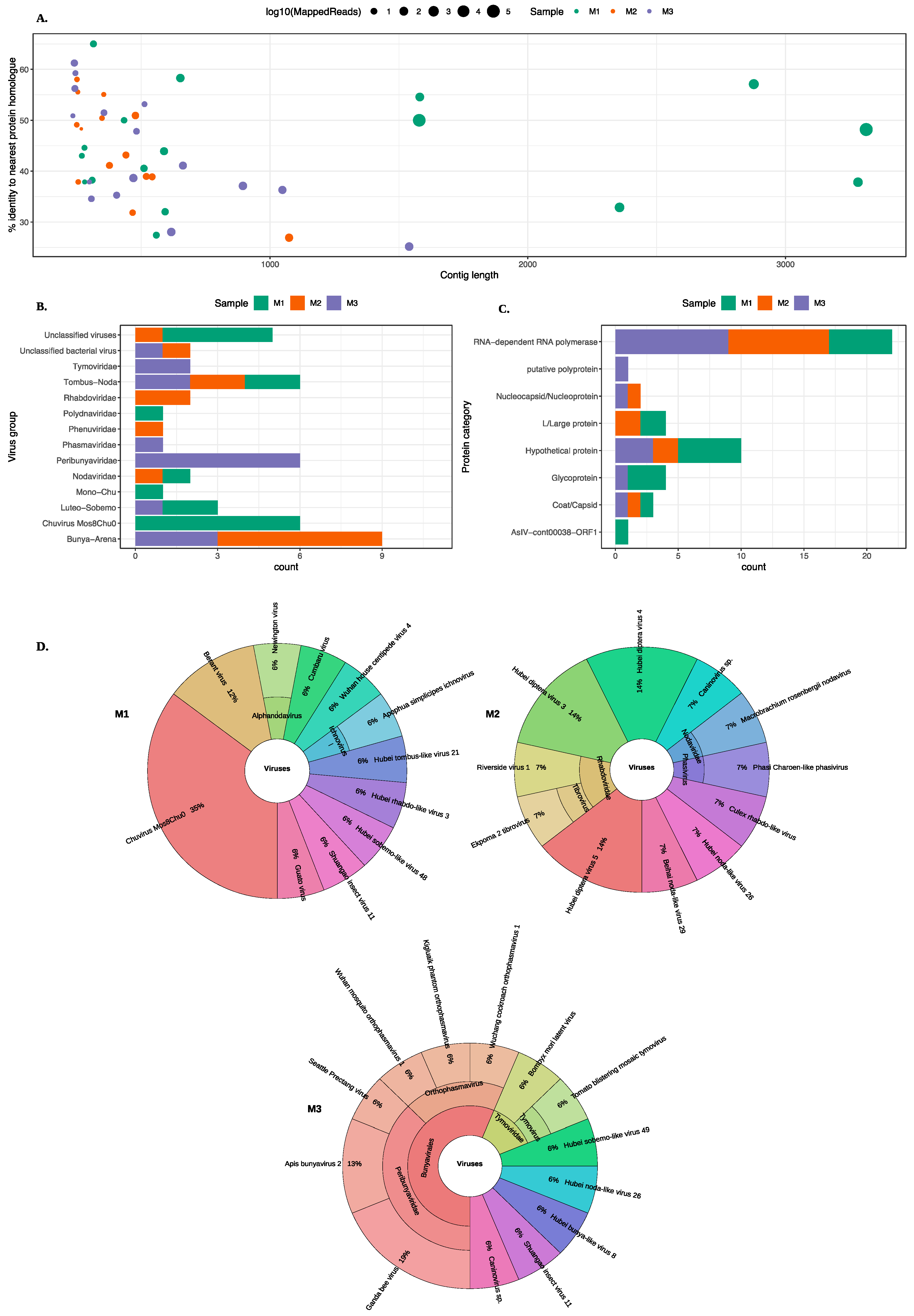
3.1. Identification of Viruses from Midge Pools
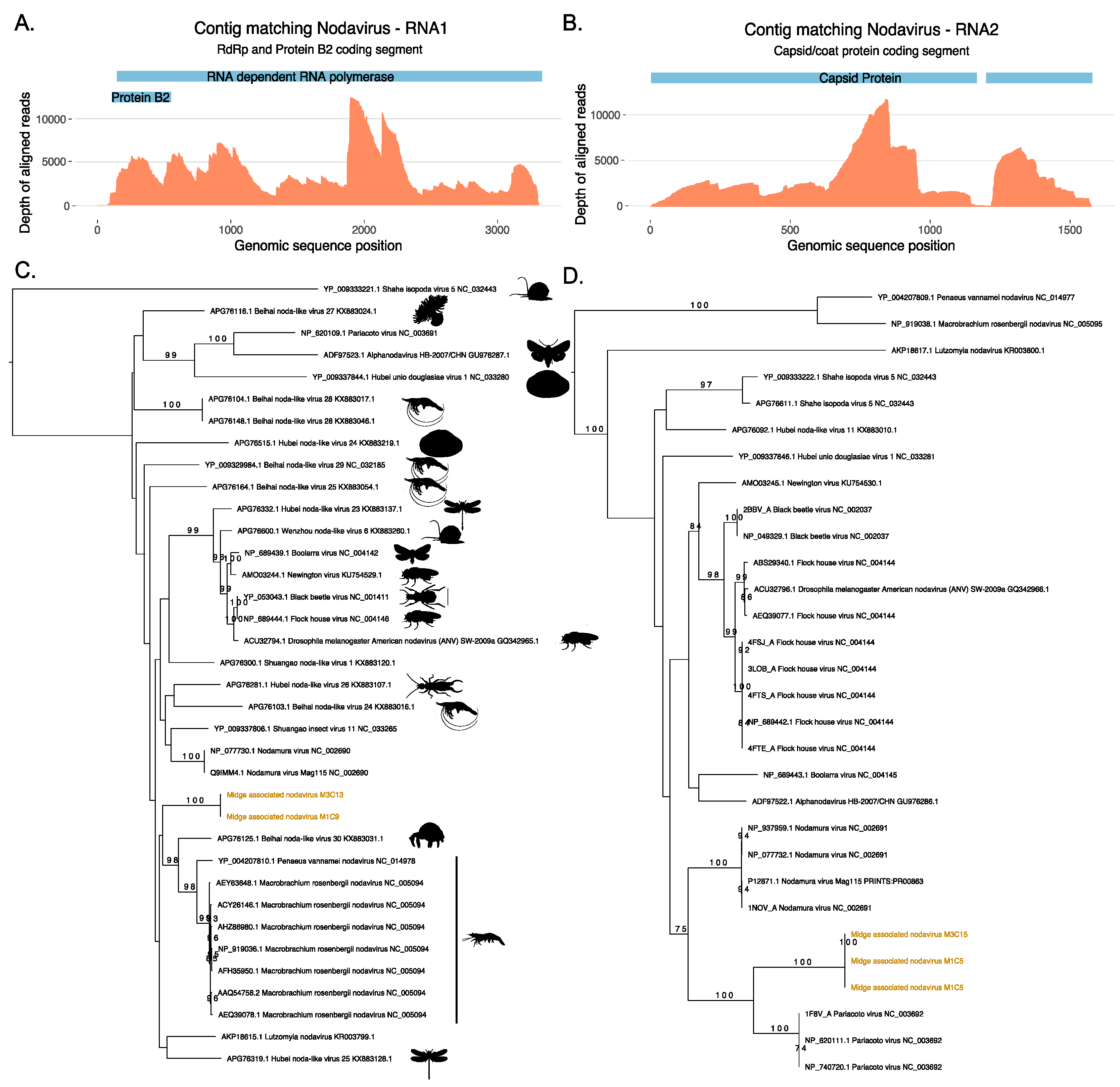
3.2. Identification of a Novel Nodavirus from Two Midge Pools
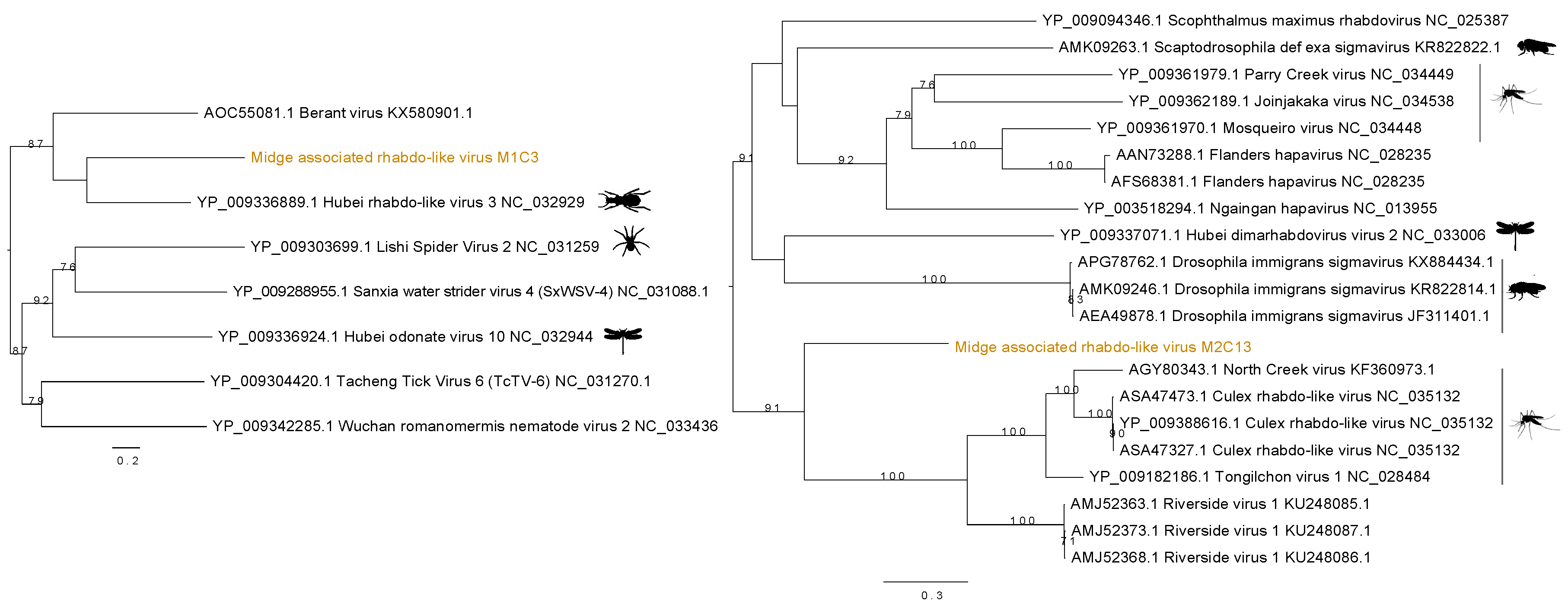
3.3. Identification of Rhabdo-Like Viruses in Midges
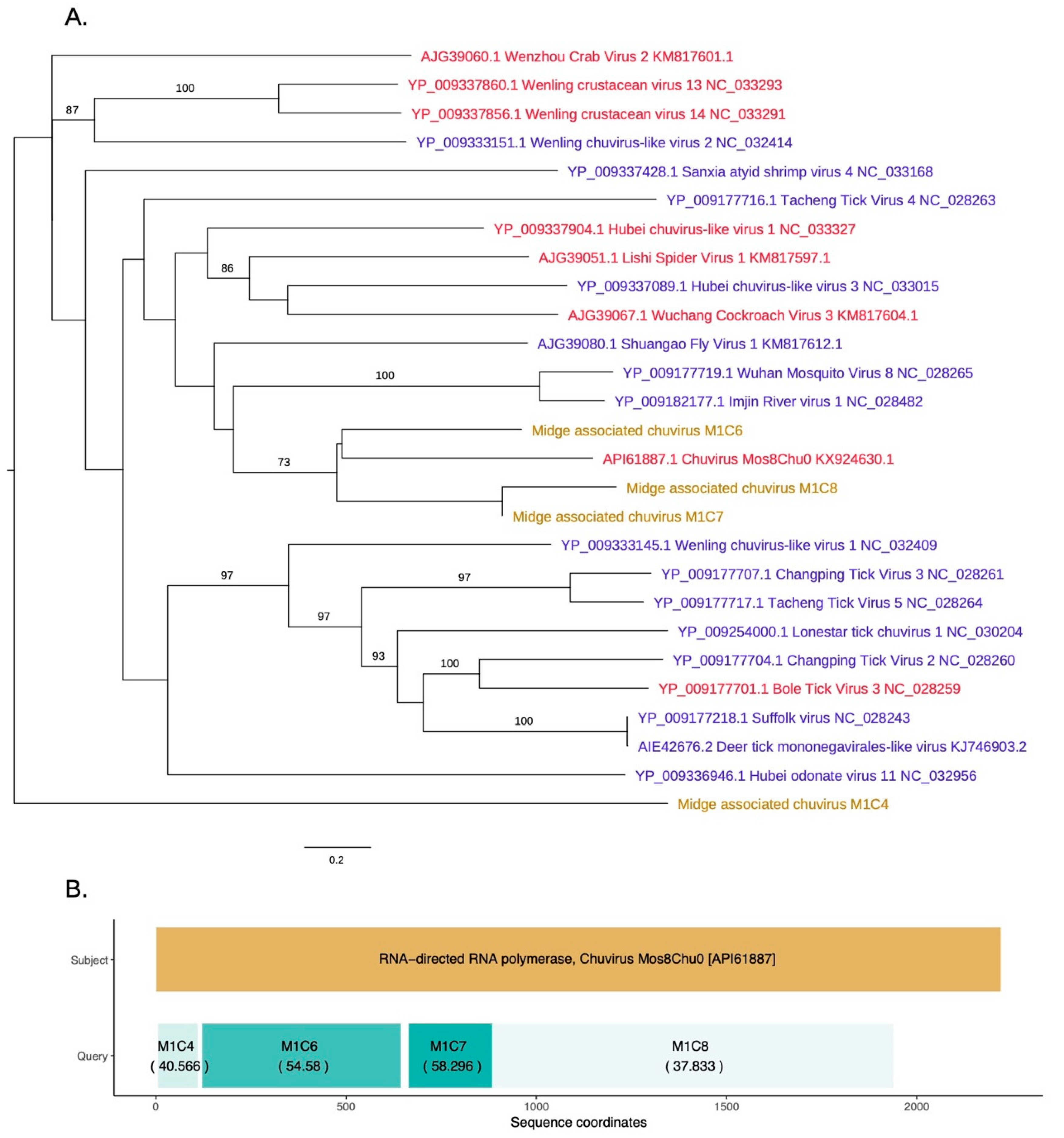
3.4. Identification of a Novel Chuvirus
3.5. Identification of Bunya-Like Viruses
3.6. Identification of Luteo-Sobemo Like Viruses
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Breitbart, M.; Rohwer, F. Here a virus, there a virus, everywhere the same virus? Trends Microbiol. 2005, 13, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Senkevich, T.G.; Dolja, V.V. The ancient Virus World and evolution of cells. Biol. Direct 2006, 1, 29. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V.; Krupovic, M. Origins and evolution of viruses of eukaryotes: The ultimate modularity. Virology 2015, 479–480, 2–25. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-X.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Kang, Y.-J.; Chen, L.-J.; Qin, X.-C.; Xu, J.; Holmes, E.C.; Zhang, Y.-Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Lin, X.-D.; Tian, J.-H.; Chen, L.-J.; Chen, X.; Li, C.-X.; Qin, X.-C.; Li, J.; Cao, J.-P.; Eden, J.-S.; et al. Redefining the invertebrate RNA virosphere. Nature 2016, 540, 539–543. [Google Scholar] [CrossRef] [PubMed]
- Delwart, E.L. Viral metagenomics. Rev. Med. Virol. 2007, 17, 115–131. [Google Scholar] [CrossRef] [PubMed]
- Nooij, S.; Schmitz, D.; Vennema, H.; Kroneman, A.; Koopmans, M.P.G. Overview of Virus Metagenomic Classification Methods and Their Biological Applications. Front. Microbiol. 2018, 9, 749. [Google Scholar] [CrossRef]
- Simmonds, P.; Adams, M.J.; Benkő, M.; Breitbart, M.; Brister, J.R.; Carstens, E.B.; Davison, A.J.; Delwart, E.; Gorbalenya, A.E.; Harrach, B.; et al. Consensus statement: Virus taxonomy in the age of metagenomics. Nat. Rev. Microbiol. 2017, 15, 161–168. [Google Scholar] [CrossRef]
- McHugh, C.P. Arthropods: Vectors of Disease Agents. Lab. Med. 1994, 25, 429–437. [Google Scholar] [CrossRef][Green Version]
- Reeves, W.C. Arthropods as Vectors and Reservoirs of Animal Pathogenic Viruses. In Handbuch der Virusforschung; Springer: Berlin, Germany, 1958; pp. 177–202. [Google Scholar]
- Roundy, C.M.; Azar, S.R.; Rossi, S.L.; Weaver, S.C.; Vasilakis, N. Insect-Specific Viruses: A Historical Overview and Recent Developments. In Advances in Virus Research; Academic Press: Cambridge, MA, USA, 2017; Volume 98, pp. 119–146. [Google Scholar]
- Weaver, S.C.; Charlier, C.; Vasilakis, N.; Lecuit, M. Zika, Chikungunya, and Other Emerging Vector-Borne Viral Diseases. Annu. Rev. Med. 2018, 69, 395–408. [Google Scholar] [CrossRef]
- Qin, X.-C.; Shi, M.; Tian, J.-H.; Lin, X.-D.; Gao, D.-Y.; He, J.-R.; Wang, J.-B.; Li, C.-X.; Kang, Y.-J.; Yu, B.; et al. A tick-borne segmented RNA virus contains genome segments derived from unsegmented viral ancestors. Proc. Natl. Acad. Sci. USA 2014, 111, 6744–6749. [Google Scholar] [CrossRef] [PubMed]
- Remnant, E.J.; Shi, M.; Buchmann, G.; Blacquière, T.; Holmes, E.C.; Beekman, M.; Ashe, A. A Diverse Range of Novel RNA Viruses in Geographically Distinct Honey Bee Populations. J. Virol. 2017, 91, e00158-17. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Neville, P.; Nicholson, J.; Eden, J.-S.; Imrie, A.; Holmes, E.C. High Resolution Meta-Transcriptomics Reveals the Ecological Dynamics of Mosquito-Associated RNA Viruses in Western Australia. J. Virol. 2017, 91, e00680-17. [Google Scholar] [CrossRef] [PubMed]
- Coffey, L.L.; Page, B.L.; Greninger, A.L.; Herring, B.L.; Russell, R.C.; Doggett, S.L.; Haniotis, J.; Wang, C.; Deng, X.; Delwart, E.L. Enhanced arbovirus surveillance with deep sequencing: Identification of novel rhabdoviruses and bunyaviruses in Australian mosquitoes. Virology 2014, 448, 146–158. [Google Scholar] [CrossRef] [PubMed]
- Fauver, J.R.; Grubaugh, N.D.; Krajacich, B.J.; Weger-Lucarelli, J.; Lakin, S.M.; Fakoli, L.S.; Bolay, F.K.; Diclaro, J.W.; Dabiré, K.R.; Foy, B.D.; et al. West African Anopheles gambiae mosquitoes harbor a taxonomically diverse virome including new insect-specific flaviviruses, mononegaviruses, and totiviruses. Virology 2016, 498, 288–299. [Google Scholar] [CrossRef] [PubMed]
- de Lara Pinto, A.Z.; Santos de Carvalho, M.; de Melo, F.L.; Ribeiro, A.L.M.; Morais Ribeiro, B.; Dezengrini Slhessarenko, R. Novel viruses in salivary glands of mosquitoes from sylvatic Cerrado, Midwestern Brazil. PLoS ONE 2017, 12, e0187429. [Google Scholar] [CrossRef] [PubMed]
- Pettersson, J.H.-O.; Shi, M.; Bohlin, J.; Eldholm, V.; Brynildsrud, O.B.; Paulsen, K.M.; Andreassen, Å.; Holmes, E.C. Characterizing the virome of Ixodes ricinus ticks from northern Europe. Sci. Rep. 2017, 7, 10870. [Google Scholar] [CrossRef] [PubMed]
- Reuter, G.; Boros, Á.; Pál, J.; Kapusinszky, B.; Delwart, E.; Pankovics, P. Detection and genome analysis of a novel (dima)rhabdovirus (Riverside virus) from Ochlerotatus sp. mosquitoes in Central Europe. Infect. Genet. Evol. 2016, 39, 336–341. [Google Scholar] [CrossRef]
- Carpenter, S.; Groschup, M.H.; Garros, C.; Felippe-Bauer, M.L.; Purse, B.V. Culicoides biting midges, arboviruses and public health in Europe. Antiviral Res. 2013, 100, 102–113. [Google Scholar] [CrossRef]
- Hoffmann, B.; Scheuch, M.; Höper, D.; Jungblut, R.; Holsteg, M.; Schirrmeier, H.; Eschbaumer, M.; Goller, K.V.; Wernike, K.; Fischer, M.; et al. Novel Orthobunyavirus in Cattle, Europe, 2011. Emerg. Infect. Dis. 2012, 18, 469–472. [Google Scholar] [CrossRef]
- Nomikou, K.; Hughes, J.; Wash, R.; Kellam, P.; Breard, E.; Zientara, S.; Palmarini, M.; Biek, R.; Mertens, P. Widespread Reassortment Shapes the Evolution and Epidemiology of Bluetongue Virus following European Invasion. PLOS Pathog. 2015, 11, e1005056. [Google Scholar] [CrossRef]
- Sakkas, H.; Bozidis, P.; Franks, A.; Papadopoulou, C.; Sakkas, H.; Bozidis, P.; Franks, A.; Papadopoulou, C. Oropouche Fever: A Review. Viruses 2018, 10, 175. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, A.; Ritchie, A.; Hillman, J.R.; Fenton, B. Scottish Crop Research Institute Annual Report 2001/2002. In Scottish Crop Research Institute Annual Report 2001/2002; Scottish Crop Research Institute: Dundee, Scotland, 2003; pp. 97–100. [Google Scholar]
- Campbell, J.A.; Pelham-Clinton, E.C. X.—A Taxonomic Review of the British Species of Culicoides Latreille (Diptera, Ceratopogonidæ). Proc. R. Soc. Edinburgh. Sect. B. Biol. 1960, 67, 181–302. [Google Scholar] [CrossRef]
- Woodford, L.; Bianco, G.; Ivanova, Y.; Dale, M.; Elmer, K.; Rae, F.; Larcombe, S.D.; Helm, B.; Ferguson, H.M.; Baldini, F. Vector species-specific association between natural Wolbachia infections and avian malaria in black fly populations. Sci. Rep. 2018, 8, 4188. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, B.; Cêtre-Sossah, C.; Garros, C.; Chavernac, D.; Balenghien, T.; Carpenter, S.; Setier-Rio, M.-L.; Vignes-Lebbe, R.; Ung, V.; Candolfi, E.; et al. Development and validation of IIKC: an interactive identification key for Culicoides (Diptera: Ceratopogonidae) females from the Western Palaearctic region. Parasit. Vectors 2012, 5, 137. [Google Scholar] [CrossRef] [PubMed]
- Modha, S. MetaViC: Virus Metagenomics Pipeline for Unknown Host or in Absence of a Host Genome. Available online: https://github.com/sejmodha/metaViC (accessed on 22 August 2016).
- Krueger, F. Trim Galore—Babraham Bioinformatics. Available online: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 1 July 2016).
- Schmieder, R.; Lim, Y.W.; Edwards, R. Identification and removal of ribosomal RNA sequences from metatranscriptomes. Bioinformatics 2012, 28, 433–435. [Google Scholar] [CrossRef] [PubMed]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glockner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef]
- Quast, C.; Pruesse, E.; Yilmaz, P.; Gerken, J.; Schweer, T.; Yarza, P.; Peplies, J.; Glöckner, F.O. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 2012, 41, D590–D596. [Google Scholar] [CrossRef]
- Glöckner, F.O.; Yilmaz, P.; Quast, C.; Gerken, J.; Beccati, A.; Ciuprina, A.; Bruns, G.; Yarza, P.; Peplies, J.; Westram, R.; et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 2017, 261, 169–176. [Google Scholar] [CrossRef]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef]
- O’Leary, N.A.; Wright, M.W.; Brister, J.R.; Ciufo, S.; Haddad, D.; McVeigh, R.; Rajput, B.; Robbertse, B.; Smith-White, B.; Ako-Adjei, D.; et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res. 2016, 44, D733–D745. [Google Scholar] [CrossRef] [PubMed]
- Schmieder, R.; Edwards, R. Quality control and preprocessing of metagenomic datasets. Bioinformatics 2011, 27, 863–864. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Chin, F.Y.L. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Smits, S.L.; Bodewes, R.; Ruiz-González, A.; Baumgärtner, W.; Koopmans, M.P.; Osterhaus, A.D.M.E.; Schürch, A.C. Recovering full-length viral genomes from metagenomes. Front. Microbiol. 2015, 6, 1069. [Google Scholar] [CrossRef]
- Soto-Jimenez, L.; Estrada, K.; Sanchez-Flores, A. GARM: Genome Assembly, Reconciliation and Merging Pipeline. Curr. Top. Med. Chem. 2014, 14, 418–424. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed]
- Vicedomini, R.; Vezzi, F.; Scalabrin, S.; Arvestad, L.; Policriti, A. GAM-NGS: Genomic assemblies merger for next generation sequencing. BMC Bioinformatics 2013, 14, S6. [Google Scholar] [CrossRef]
- Wences, A.H.; Schatz, M.C. Metassembler: Merging and optimizing de novo genome assemblies. Genome Biol. 2015, 16, 207. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Gurevich, A.; Saveliev, V.; Vyahhi, N.; Tesler, G. QUAST: Quality assessment tool for genome assemblies. Bioinformatics 2013, 29, 1072–1075. [Google Scholar] [CrossRef] [PubMed]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinform. 2011, 12, 385. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Jones, P.; Binns, D.; Chang, H.-Y.; Fraser, M.; Li, W.; McAnulla, C.; McWilliam, H.; Maslen, J.; Mitchell, A.; Nuka, G.; et al. InterProScan 5: Genome-scale protein function classification. Bioinformatics 2014, 30, 1236–1240. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Suyama, M.; Torrents, D.; Bork, P. PAL2NAL: Robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 2006, 34, W609–W612. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Sahul Hameed, A.S.; Ninawe, A.S.; Nakai, T.; Chi, S.C.; Johnson, K.L.; Consortium, I.R. ICTV Virus Taxonomy Profile: Nodaviridae. J. Gen. Virol. 2019, 100, 3–4. [Google Scholar] [CrossRef]
- Öhlund, P.; Lundén, H.; Blomström, A.-L. Insect-specific virus evolution and potential effects on vector competence. Virus Genes 2019, 55, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Hendry, G.A.F.; Godwin, G. Biting midges in Scottish forestry: A costly irritant or a trivial nuisance? Scottish For. 1988, 42, 113–119. [Google Scholar]
- Žiegytė, R.; Markovets, M.Y.; Bernotienė, R.; Mukhin, A.; Iezhova, T.A.; Valkiūnas, G.; Palinauskas, V. The widespread biting midge Culicoides impunctatus (Ceratopogonidae) is susceptible to infection with numerous Haemoproteus (Haemoproteidae) species. Parasit. Vectors 2017, 10, 397. [Google Scholar] [CrossRef] [PubMed]
- Longdon, B.; Murray, G.G.R.; Palmer, W.J.; Day, J.P.; Parker, D.J.; Welch, J.J.; Obbard, D.J.; Jiggins, F.M. The evolution, diversity, and host associations of rhabdoviruses. Virus Evol. 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Stork, N.E. How Many Species of Insects and Other Terrestrial Arthropods Are There on Earth? Annu. Rev. Entomol. 2018, 63, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Modha, S.; Thanki, A.; Cotmore, S.F.; Davison, A.J.; Hughes, J.; Kelso, J. ViCTree: An automated framework for taxonomic classification from protein sequences. Bioinformatics 2018, 1, 6. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Modha, S.; Hughes, J.; Bianco, G.; Ferguson, H.M.; Helm, B.; Tong, L.; Wilkie, G.S.; Kohl, A.; Schnettler, E. Metaviromics Reveals Unknown Viral Diversity in the Biting Midge Culicoides impunctatus. Viruses 2019, 11, 865. https://doi.org/10.3390/v11090865
Modha S, Hughes J, Bianco G, Ferguson HM, Helm B, Tong L, Wilkie GS, Kohl A, Schnettler E. Metaviromics Reveals Unknown Viral Diversity in the Biting Midge Culicoides impunctatus. Viruses. 2019; 11(9):865. https://doi.org/10.3390/v11090865
Chicago/Turabian StyleModha, Sejal, Joseph Hughes, Giovanni Bianco, Heather M. Ferguson, Barbara Helm, Lily Tong, Gavin S. Wilkie, Alain Kohl, and Esther Schnettler. 2019. "Metaviromics Reveals Unknown Viral Diversity in the Biting Midge Culicoides impunctatus" Viruses 11, no. 9: 865. https://doi.org/10.3390/v11090865
APA StyleModha, S., Hughes, J., Bianco, G., Ferguson, H. M., Helm, B., Tong, L., Wilkie, G. S., Kohl, A., & Schnettler, E. (2019). Metaviromics Reveals Unknown Viral Diversity in the Biting Midge Culicoides impunctatus. Viruses, 11(9), 865. https://doi.org/10.3390/v11090865