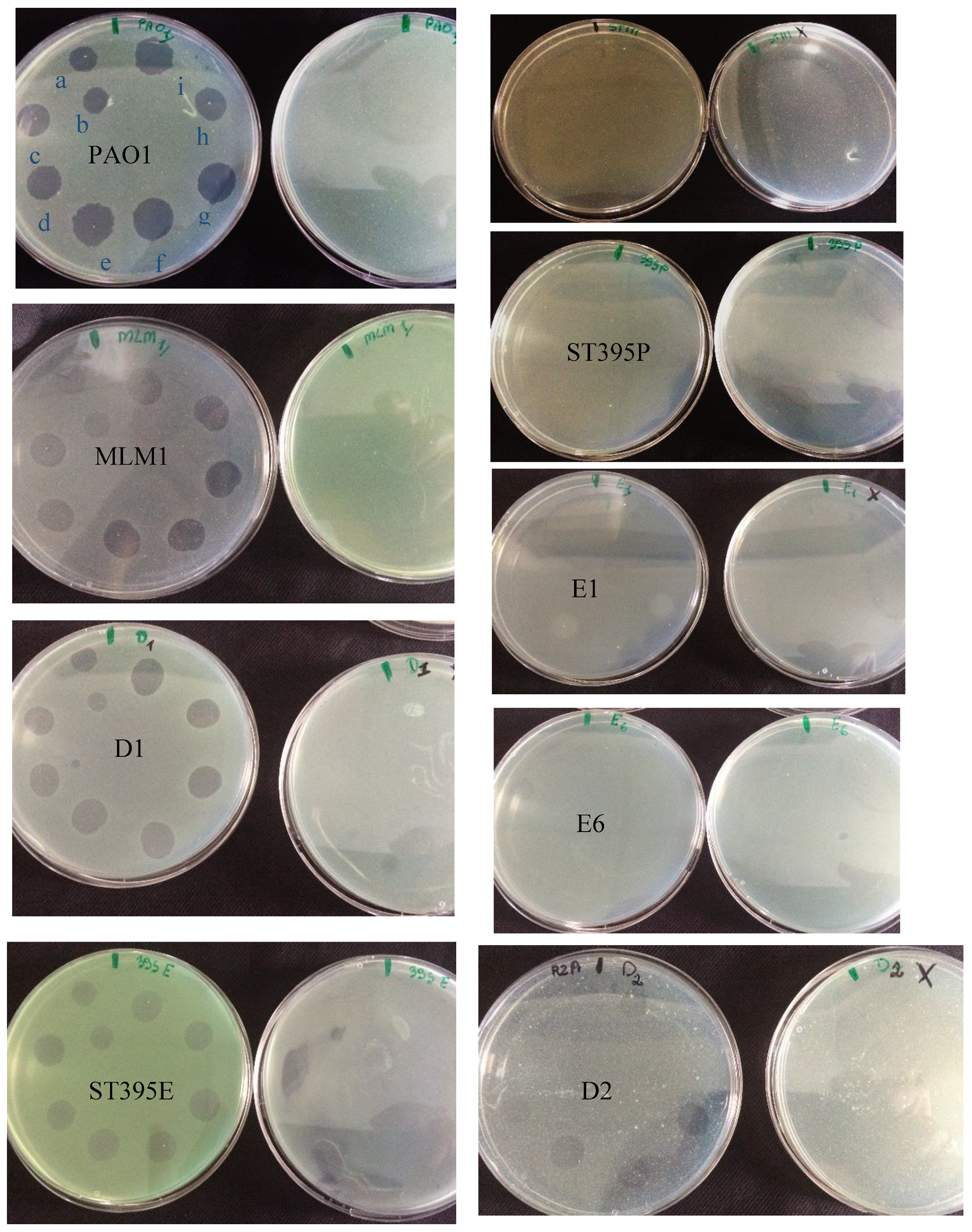
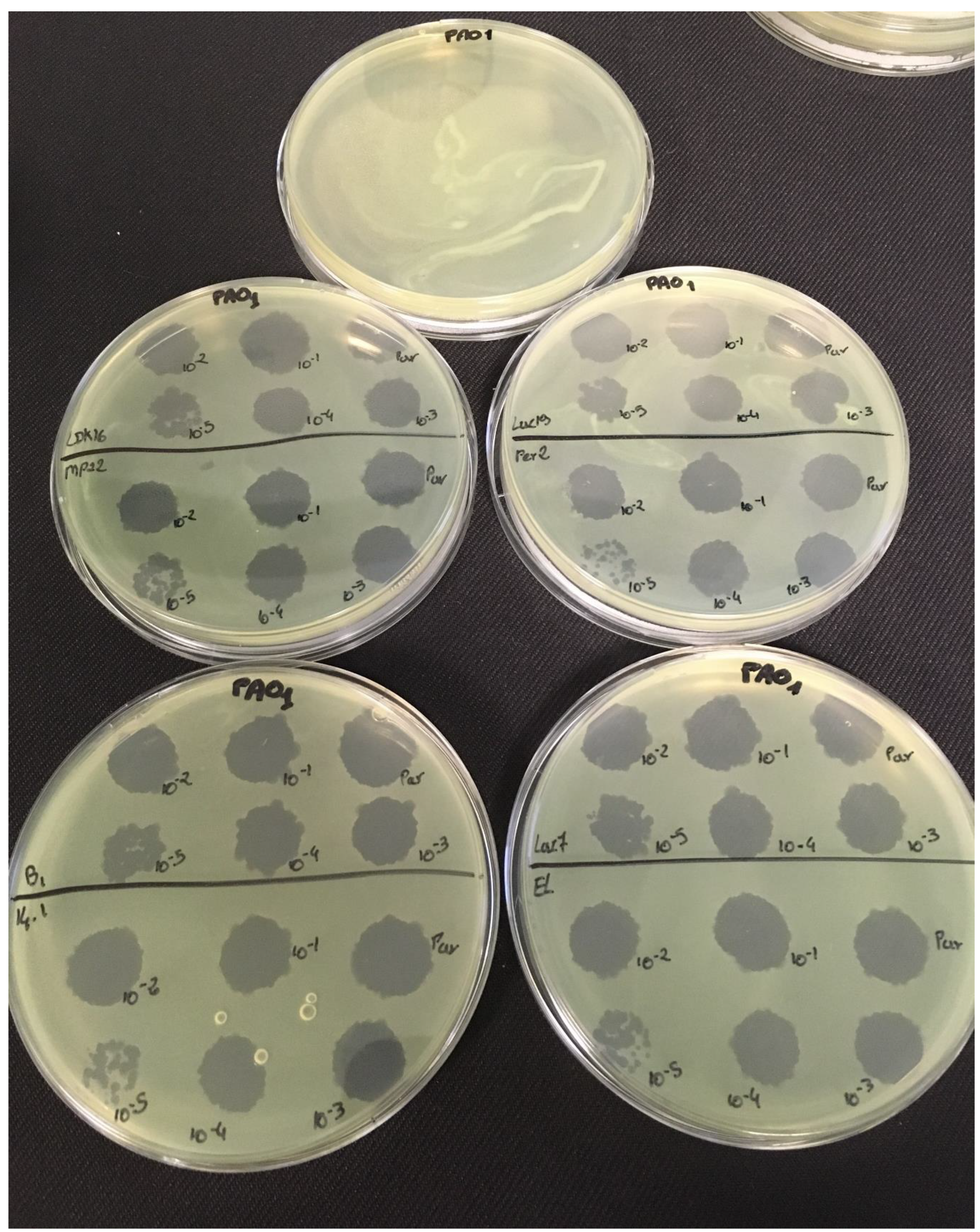
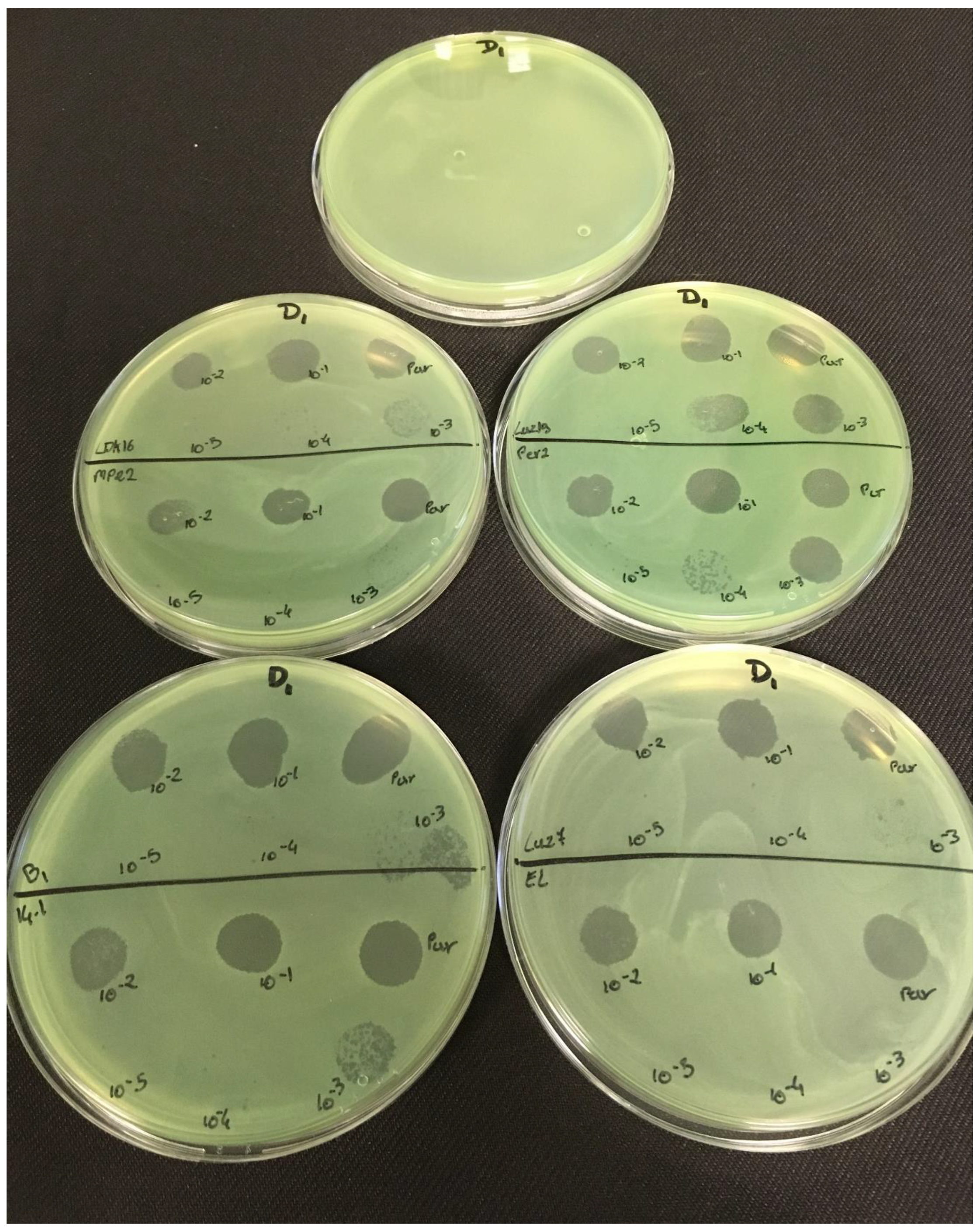
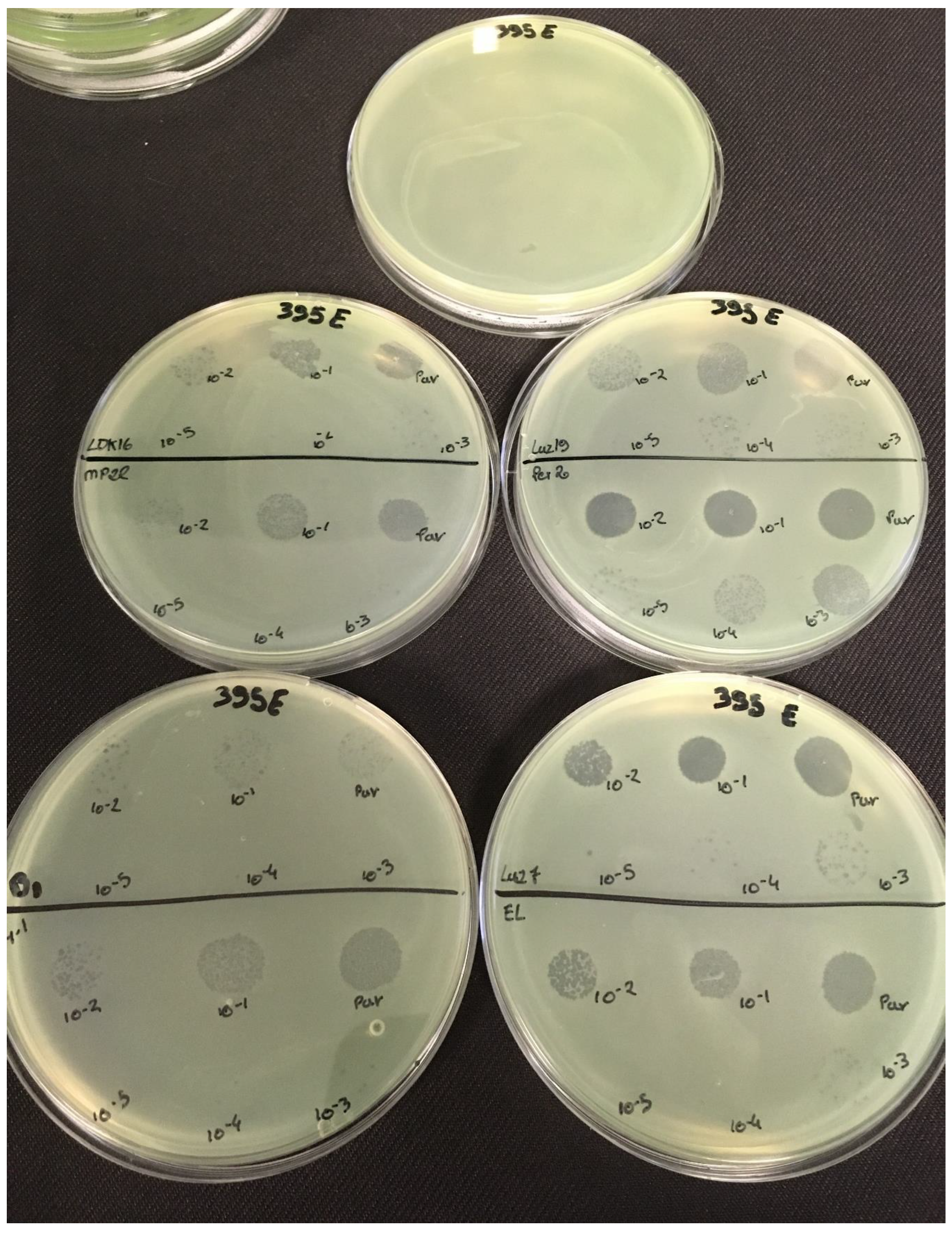
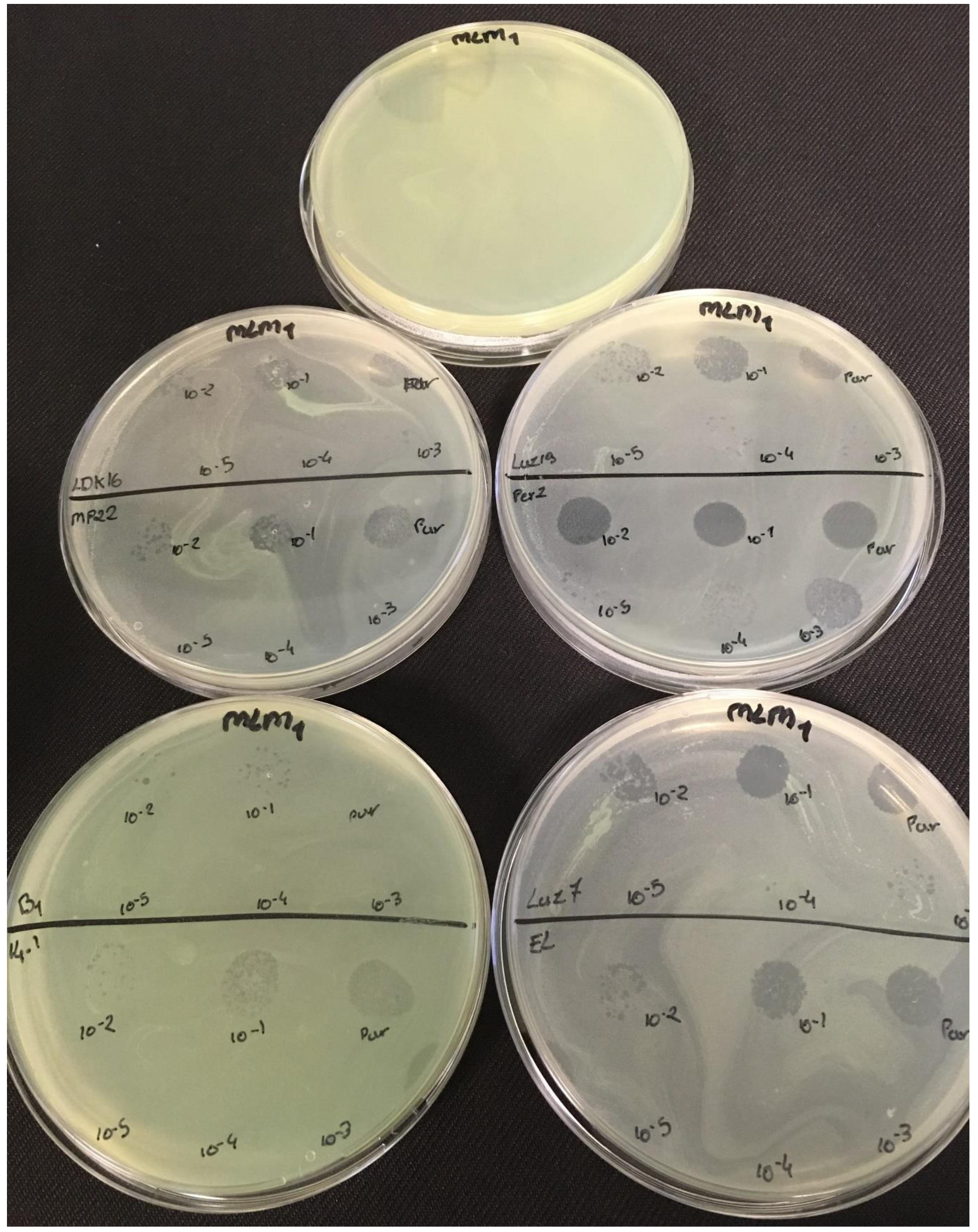
3.2. Effectiveness of Individual Phage: Selection of Virulent Phages
Based on previous experiments, four
P. aeruginosa strains were selected (MLM, ST395E, PA01 and D1). These strains were challenged with all phages to estimate the effectiveness of each one according to the strain. The spot test method was also used, but standardized phage solutions (10
9 PFU/mL) were diluted from 10
−1 to 10
−5 before being spotted onto the agar lawn. The minimum infectious titer (MIT) was determined for each infectious phage. Results from
Figure A2,
Figure A3,
Figure A4 and
Figure A5 are summarized in
Table 5 and were used to select three viruses.
The trials showed the same results for the eight phages concerning the PAO1 strain with the smallest MIT (106 PFU/mL). The LUZ7, PEV2, and 14.1 bacteriophages were able to efficiently lyse the MLM and ST395E strains at the same titers, for example, 107 PFU/ mL for LUZ7 and PEV2 and 109 PFU/mL for 14.1. All phages were infectious on the D1 strain, and most efficient ones were LUZ7, LUZ19, PEV2, B1, 14.1, and LDK16 with the MIT estimated at 107 PFU/mL. Based on these results, LUZ7 (Podoviridae) and 14.1 (Myoviridae) were retained for their effectiveness and in order to represent two of the three Caudovirales families. The newly isolated B1 phage was the third candidate selected to assess the susceptibility of P. aeruginosa planktonic cells from the MLM, D1, and PAO1 strains.
3.3. Observation of B1 Phages
Transmission electron microscopy was used to observe B1 viral particles. The 14.1 and LUZ7 phages were used as references. Observations show particles with a head and tail measuring approximately 100 nm in length, confirming that phage 14.1 is a member of
Myoviridae (
Figure 2A), which is in accordance with the literature. The PB1 family, to which 14.1 belongs, brings together phages with an icosahedral head of about 70 nm and a non-striated tail of about 50 nm [
45]. Pictures taken on the same grid also allowed phages without a tail to be observed (
Figure 2A). Similar structures in the surrounding environment suggest that during the resuspension of phages in solution by vortexing before depositing them on the grid, some phages could have been damaged by losing their appendage to leave an outgrowth (see arrow).
This alteration of particles would be a source of bias during TEM characterization [
65]. It also suggests that if
Podoviridae are less affected, 15% to 40% of
Myoviridae and between 32% and 76% of
Siphoviridae can be altered during extraction.
In the case of LUZ7 phages, observations made on the purified extracts show structures with hexagonal forms without tails, measuring approximately 50 to 60 nm, suggesting that they are members of the
Podoviridae family (
Figure 2B). In the literature, LUZ7 assimilated to the family of N4 viruses [
46] is also described as a Podoviridae with an icosahedral head of 70 nm. Observations made by Ceyssens et al., however, do not allow the tail to be observed, which, in this family, is considered to be less than 30 nm. In some cases, uranyl ions have a strong affinity with double-stranded DNA, which causes a black color inside the capsid. Similar observations have already been made by E. Bradley [
66] in a review dedicated to the study of phage morphology by microscopy. Ackermann and Tiekotter [
67] also spoke of positive staining. This artifact is, in most cases, the cause of an underestimation of the size of a viral particle of about 10% to 15%, and does not allow the secondary structures such as the tail or the capsomeres to be observed [
67].
Pictures of the B1 unknown extract (
Figure 2C) show particles of homogeneous and icosahedral shape of about 60 nm. The absence of a visible caudal appendage during the observations is in favor of viral particles probably belonging to
Podoviridae.
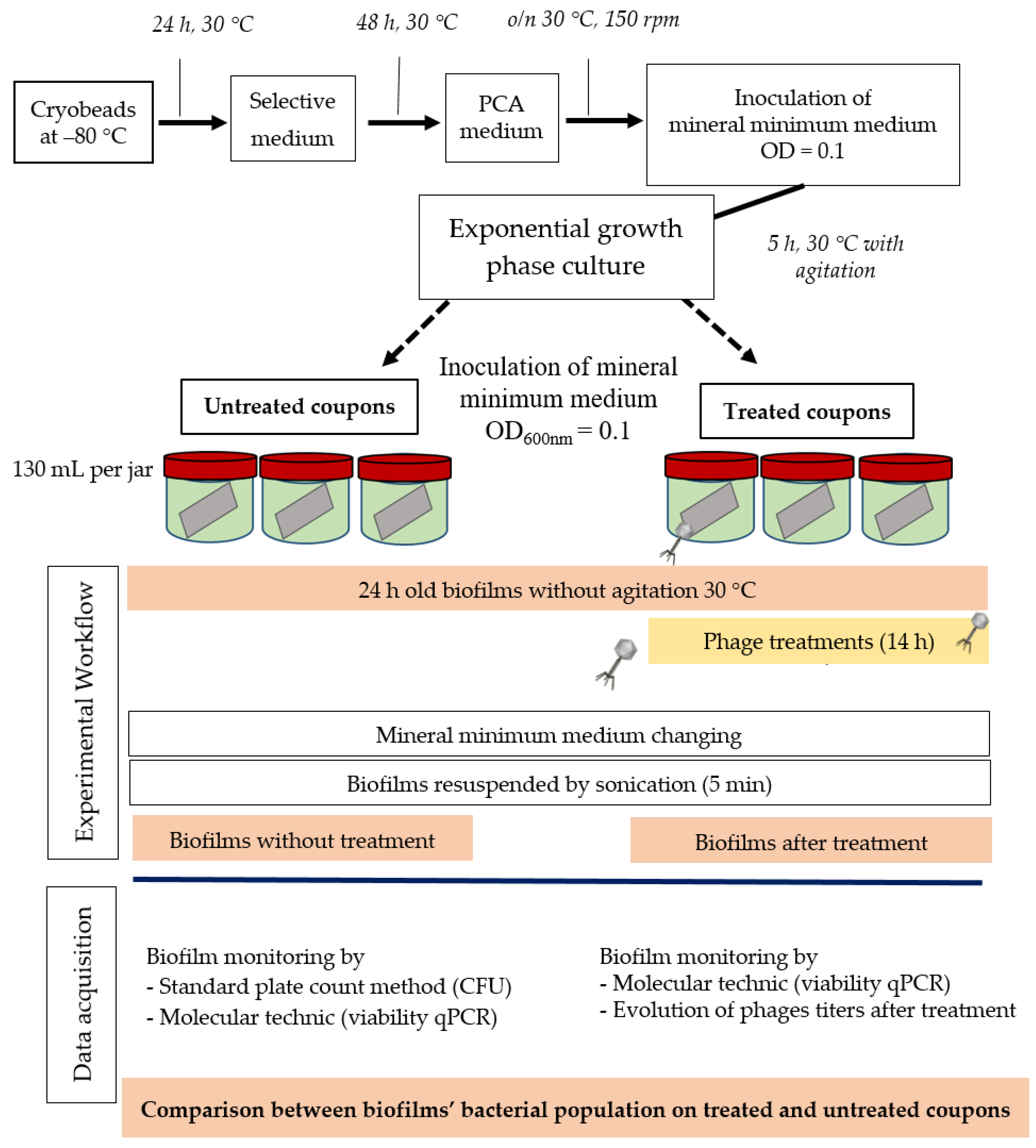
3.5. Liquid Infectious Pattern of LUZ7, B1, and 14.1 in Mineral Minimum Medium
The efficacy of the three phages, selected above, was assessed by inoculating a culture of P. aeruginosa PAO1 and also the three sensitive strains MLM, D1, and ST395E. Suspensions containing the phages were prepared in mineral minimum medium to achieve a multiplicity of infection (MOI, the ratio of virus particles to viable bacterial cells) of 0.1/1/10 in the culture. Phages were introduced after 14 h of bacterial growth, and then cultures were monitored for a further 24 h period. The activity of phages significantly reduced the bacterial population, but the magnitude of these reductions was strain-dependent.
Figure 3,
Figure 4 and
Figure 5 show that for all controls without phages, absorbance was two-fold higher after 24 h. Conversely, when bacteriophages were infectious, OD
600 was decreased to near zero. In the case of LUZ7, two patterns were underlined (
Figure 3). The populations of the D1 and PAO1 strains were decreased by 90% and 95% respectively, from 8 to 24 h after phage inoculation. However, the MOI which initiated the decline after about 2 h of time course, was the highest. Surprisingly, LUZ7, although selected on the basis of spot tests, did not suppress the growth of the host strains MLM and ST395E at all MOIs tested.
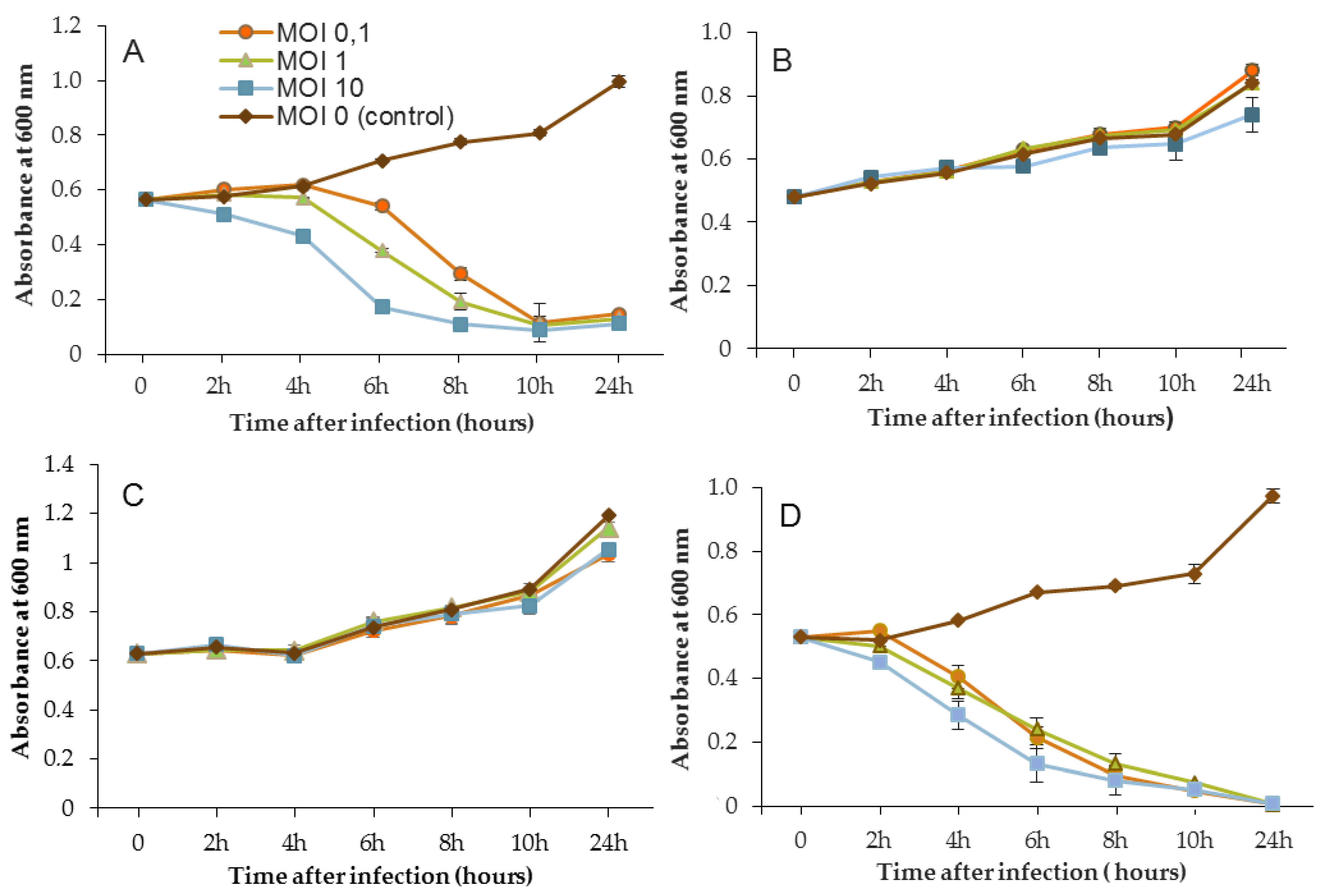
In the same way, the growth of D1 and PAO1 strains was stopped and then decreased from 2 h after inoculation of phage 14.1 for the highest MOI (
Figure 4). Other MOIs (1 and 0.1) also had an effect, but this was delayed in time. Nonetheless, all of the MOIs tested led to the same rate of growth inhibition (approximately 90%) at 10 h post-inoculation. Although the antibacterial effect of phages was significant on the PAO1 and D1 strains, populations seemed to behave differently between 10 and 24 h. Data led to thinking about potential regrowth.
Compared to LUZ7, the phage 14.1 (at MOI 10) was able to maintain the steady state of the MLM and ST395E populations but not along the whole time course of the infection assay (
Figure 4), because after 10 h, the optical density values increased.
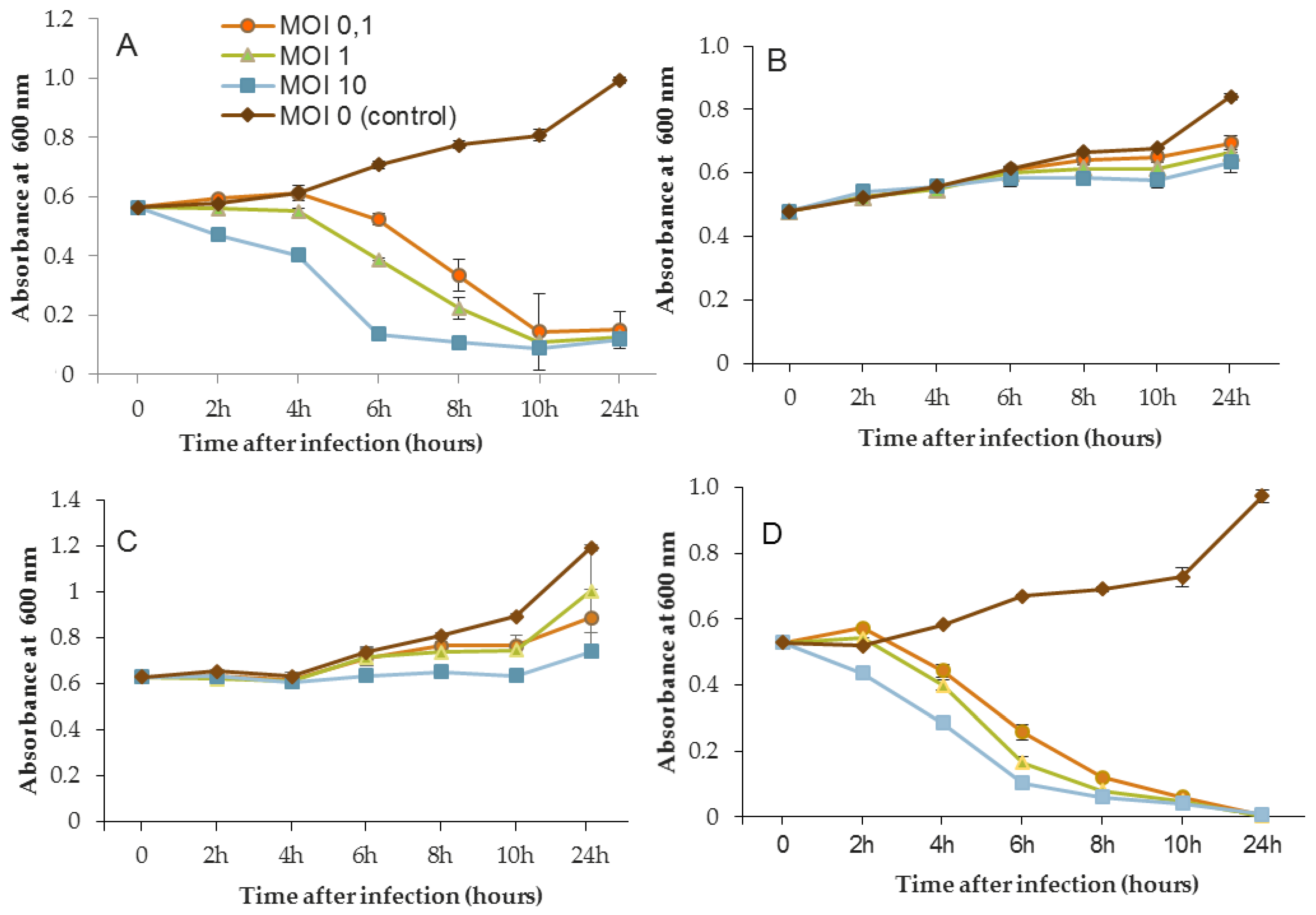
The phage activity of B1 was very different from that of LUZ7 and 14.1 (
Figure 5). If they had the same infection pattern against the PAO1 strain and D1 isolate, the results showed that B1 could interfere with ST35E and MLM planktonic cells. Concerning the MLM and ST395 cultures, OD
600 values evolved in the same manner as controls without phages during the first 8 h of phage treatment (
Figure 5B,C) and then, surprisingly, phage B1 (at MOI 10) managed to negatively impact the total population, which was reduced by 45% and 70%, respectively (
Figure 5B,C).
Planktonic cells from the reference PAO1 strain, known as a permissive strain, were efficiently eradicated by each of the three viruses, LUZ7, 14.1 and B1 (
Figure 3D,
Figure 4D and
Figure 5D), in liquid. The environmental strain D1 was also sensitive to LUZ7, 14.1 and B1 (
Figure 3A,
Figure 4A and
Figure 5A). While the remaining absorbance for PAO1 could be due to bacterial components of cellular debris, the results show that the regrowth of other strains can be observed more or less rapidly after phage application. This especially concerned MLM and ST35E isolates when infected by LUZ7 and 14.1 (
Figure 3B,C and
Figure 4B,C). Infections did not suppress multiplication of the bacterial population; nonetheless, their lytic activity maintained a steady state which was probably due to equal lytic and growth rates. However, when the cell multiplication of cultures became higher than the number of dead cells, the optical density proportionally increased according to the MOI used. In the case of 14.1, the growth remained, however, at a lower rate compared to the hosts cell control grown in the absence of bacteriophages (
Figure 4B,C). Additional assays were performed by adding a second dose of the virus. No supplemental bactericidal effects were obtained (data not shown) suggesting the selection and multiplication of non-susceptible cells. Similar trends were also observed in earlier studies for
P. aeruginosa infected with
Podoviridae or
Myoviridae phages [
20,
26] and were associated with the emergence of phage-resistant cells. These phases of steady state bacteria–virus interaction can, to some extent, select natural resistant bacteria or provide the evolution of host cells by developing molecular mechanisms of resistance to bacteriophage infection. Amongst them, Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR), restriction-modification, abortive infection, or the mutation of surface receptors are the most encountered [
68]. More recently, the BREX (Bacteriophage Exclusion) system was also described as a system of defense that relies on methylation [
69].
One of the most important parameters concerns the applied infectious dose. The three MOIs tested in this study led to the same results after 24 h of phage treatment depending on the strain; however, the highest MOI sped up the elimination of bacteria.
Another point of discussion is the lack robustness of the spot test method when screening the lytic activity of phages. Pires et al. [
70] also pointed out this aspect in their study. Despite the broad spectra of activity of the four selected phages against clinical
P. aeruginosa isolates, infection assays on planktonic cells showed that two of them failed to infect their hosts, even at the ideal exponential growth phase. This evidence indicates that phages used for specific antibacterial applications should not be chosen solely on the basis of spot assays. Recently, Xie et al. [
71] proposed a microtiter plate-based assay for the determination of the bacteriophage host range and virulence in a high-throughput, 96-well format. This real-time measurement of optical density in the presence of phages would be an alternative host range method with high resolution compared to conventional agar overlay spot assays.
The viruses LUZ7 and 14.1, already known, were chosen to establish the model of the biofilms’ infection. Their effectiveness on D1 and PAO1 planktonic cells makes them good candidates to evaluate their potential on the more complex biofilm structures of these two isolates, respectively.
3.6. Activity of 14.1 and LUZ7 on Biofilms
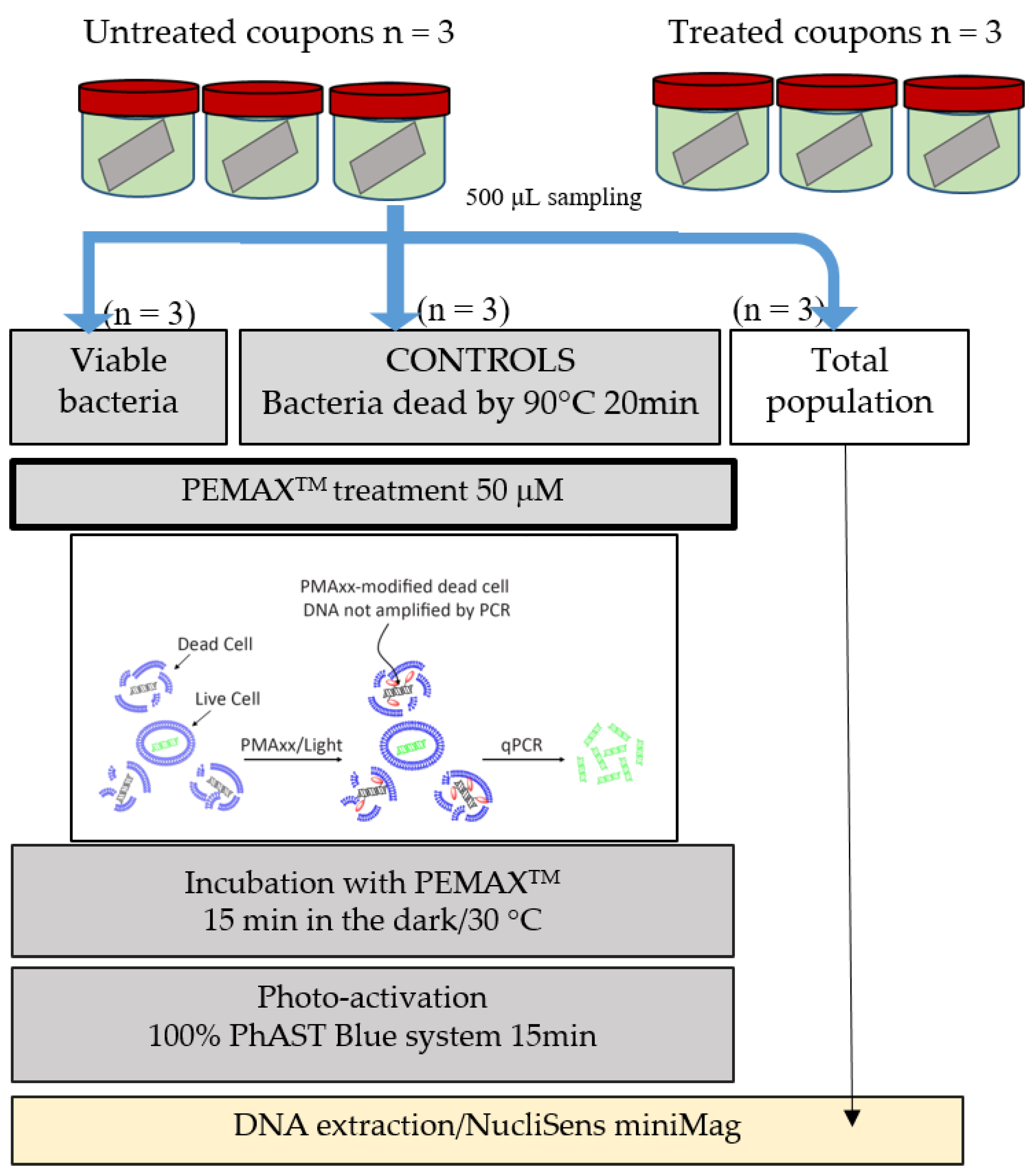
The following step was to characterize the efficiency of the bacteriophages LUZ7 and 14.1 to remove cells attached to stainless steel surfaces. Previous studies have already highlighted that the antibacterial effectiveness of phages depends partly on the host cells’ physiological state related to the biofilm formation circumstances and, on the other hand, the importance of phage infection modalities [
72,
73]. Here, juvenile, 24 h old biofilms of D1 and PAO1
P. aeruginosa were established on stainless steel coupons immersed in mineral minimum medium. Bacterial populations were quantified by standard plate count (SPC) and/or qPCR/viability qPCR (v-qPCR) before virus application and after. v-qPCR is an accurate specific method of amplification, providing the relative quantification of viable cells only (including VBNC cells) compared to the q-PCR standard, which non-selectively amplifies the targeted sequences of all bacteria (dead, viable, and VBNC) [
74,
75]. Therefore, the difference between these two types of data is one way to gain information about the repartition state of metabolic cells (between dead cells and viable + VBNC cells). Previously, the use of v-qPCR was also applied to monitor the population dynamics of five species in oral biofilms [
76], to quantify
P. aeruginosa in mono- and multispecies biofilms following exposure to various antibiotics used to treat cystic fibrosis [
60], and recently, to monitor viable cells of the biological control agent
Lactobacillus plantarum PM411 on aerial plant surfaces [
61].
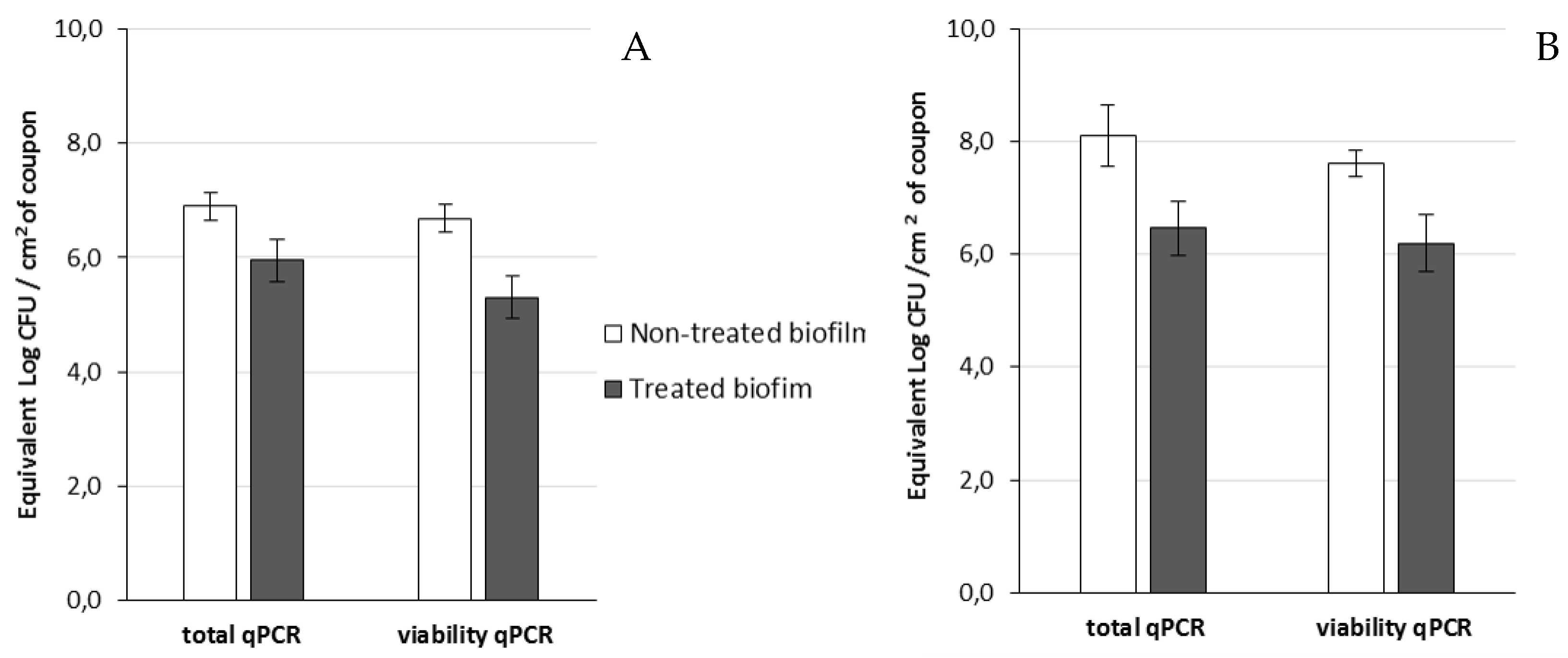
The viable SPC, qPCR, and v-qPCR results for 24 h old biofilms were respectively 7.40, 7.31 ± 0.23, and 7.12 ± 0.33 Log UFC/cm
2 of the coupon for PAO1 and 8.20, 7.44 ± 0.4, and 7.18 ± 0.39 Log CFU/cm
2 of the coupon for D1 (
Table 7). Data obtained by qPCR and v-qPCR were not significantly different in the two cases and suggested a homogenous physiological state of cells within the juvenile biofilm. The SPC data were identical to the qPCR results and confirmed that they were mainly composed of viable cells. SPC was also applied to enumerate viable cells of the 24 h + 14 h biofilm, which corresponded to the untreated biofilms. The results show that the populations of the 24 h and 24 h + 14 h biofilms were not significantly different (
Table 7 and
Table 8). Pires et al. [
70] also showed that the quantity of cells in a 24 h old biofilm of the PAO1 strain, growing in 24-well microplates, was identical to that of a 48 h biofilm and was close to 8 Log CFU/cm
2.
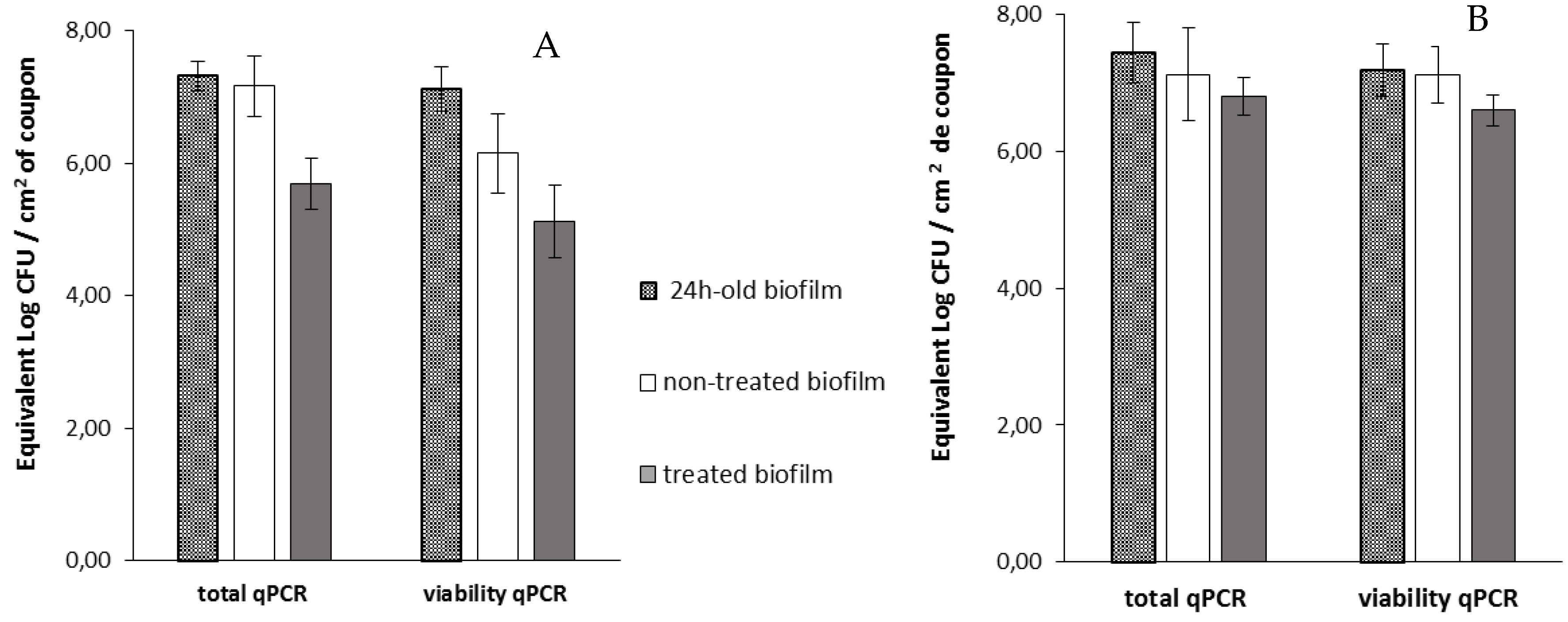
Then, viable cells were assessed 14 h after the addition of phage 14.1 or LUZ7 against PAO1 and D1 24 h-old biofilms by v-qPCR. Data were then compared to the non-treated controls that received no phage, (
Figure 6 and
Figure 7) (
Table 8). The four histograms also showed that there is no significant difference between the standard qPCR and v-qPCR results, for either treated or untreated biofilms. That means that all cells embedded to the biofilm matrix were metabolically active, infected or not. This result suggests that the application of phages in the biofilm environment does not impair the physiological state of all cells, but rather acts directly by killing and eliminating the most accessible cells. When 24 h old
P. aeruginosa PAO1 biofilms were exposed to LUZ7, the number of adhered bacteria decreased by approximately 1.5 Log (
Figure 6A) and with 14.1, the number decreased by 1.2 Log equivalent CFU/cm
2 of the coupon (
Figure 7A).The exposure of 24 h old D1 contamination led to a reduction of 0.72 Log with LUZ7 (
Figure 6B) and a reduction of 1.7 Log equivalent CFU/cm
2 of coupon with 14.1 (
Figure 7B).
Collectively, LUZ7 treatment was the most effective against the PAO1 strain compared to 14.1. This was in accordance with the short latent period and the high burst size of LUZ7 previously characterized on PAO1. On the contrary, phage 14.1 was the most effective on the 24 h old D1 biofilm. Comparing the effect of phages against free cells on biofilm removal, a more positive and rapid effect was observed in eliminating the bacterial load of broth culture than for biofilm. However, observations made over the course of experiment enabled us to make the hypothesis that phage repartition in the system could be an explanation. Indeed, the turbidity of the liquid mineral minimum medium around stainless steel coupons (INOX 316L) was totally different between the control jar without phages and the other jars (
Figure 8).
To track the distribution of viruses in the studied system, the amount of phage present in the medium around the coupons after treatment with 14.1 was determined, as well as the amount associated with the biofilm. In the assays carried out with phage 14.1, an infectious dose equivalent to 4.1 × 10
10 PFU was added to each jar. After 14 h, 6.3 × 10
11 PFU and 5 × 10
10 PFU in suspension was counted for the PAO1 strain and D1, respectively. This fraction only represents the phages which, at the end of treatment, were still completely free and virulent in the medium, since during the extraction protocol, part of the viruses adsorbed on the bacterial receptors was probably eliminated within the bacterial cell debris. The results also revealed the existence of a phage population adhered to the biofilm (10
8 PFU). This population was lower than that found in the liquid medium, although it may have been underestimated if we consider the negative impact of sonication on the viability of phage particles highlighted by Melo et al. [
77] and estimated in this study at 1 Log. LUZ7 phages showed similar behavior to a larger proportion of free-flowing phages in the medium than to associated phages (
Table 9).
This unequal distribution of phages between the liquid and solid phases is in line with the spectrophotometry results, which demonstrated, during tests with LUZ7 phages, a very significant lysis of the planktonic population in the surrounding medium (
Figure 8). In treated jars, negative values confirmed the absence of planktonic bacteria. On the basis of these observations, it is possible to discuss the mechanisms of action of bacteriophages by suggesting that:
- -
Planktonic phages exerted lytic action on the free cells detached from the biofilm as a priority, allowing them to multiply in the mineral minimum medium around the coupons.
- -
Some of these phages also adhered to the biofilm, thereby interfering with the development, as well as the spread of the biofilm in the surrounding environment.
Among the parameters likely to influence the efficacy of phages, the infectious dose used for the treatment is an important factor. By quantification of the 24 h biofilm cells, the MOI was estimated at 50 for the PAO1 strain and between 50 and 300 for the D1 strain in the case of the LUZ7 assays. These concentrations, which were much higher than those applied on the planktonic cell cultures (MOI 0.1; 1; 10), revealing the complexity of an attack targeting the biofilm. In the literature, Hanlon et al. [
78] also demonstrated the greater susceptibility of the suspended cells to phages to the detriment of the adhered cells. In their experiments, an MOI of 100 resulted in a 1-log reduction in the viable cell number of the biofilm, and the log reduction factor increased to 2 with an MOI of 1000, without the age of the biofilm mattering. Indeed, the architecture and composition of the biofilm do not facilitate access to phages [
78,
79]. The superposition of several layers of cells causes an unequal distribution of nutrients and oxygen and promotes the appearance of bacterial sub-populations whose metabolism is changed. These modifications may also impact the expression of certain genes and thus the concentration and/or the presence of the receptors used by phages during infection on the cells’ surfaces [
80,
81].
Another parameter concerns the environment-promoting cells adhesion and biofilm’s growth, as well as the kind of surface present. Phages which multiply well under in vitro conditions can fail to replicate during treatment in vivo [
25,
69]. This means that all systems should be investigated before considering phages as potential green biocides. Garbe et al. [
82] indeed showed a different behavior of the PAO1 strain cultivated in LB medium and in artificial sputum medium. Phage JG024 (
Myoviridae, PB1-Like) was therefore less effective on biofilm. In the study of Scarascia et al. [
38], three bacteriophages were further applied against a
P. aeruginosa-enriched artificial seawater biofilm to validate their activity in saline condition. One previous study also highlighted that the ability of environmental
P. aeruginosa isolates to form biofilm is correlated with the minerality of water without disturbing the metabolic activity rate [
42]. Low minerality favors biofilm production. The use of mineral minimum medium, in our study, which was composed of mineral water with a lower minerality, could thus contribute to phage progression being limited through a high-density matrix but was well representative of the contaminations found on surfaces in contact with water. The ability of
P. aeruginosa isolates to form biofilm has also been linked to the synthesis of pyocianin (PCN) [
42], a virulence factor which can generate reactive oxygen species such as H
2O
2. Some authors have suggested that it is responsible for phage inactivation [
25], but this requires further studies according the phages used.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








