Hepatitis E Virus Replication



{kind=link}
Abstract
1. Introduction
2. Genomic Organization of ORF1
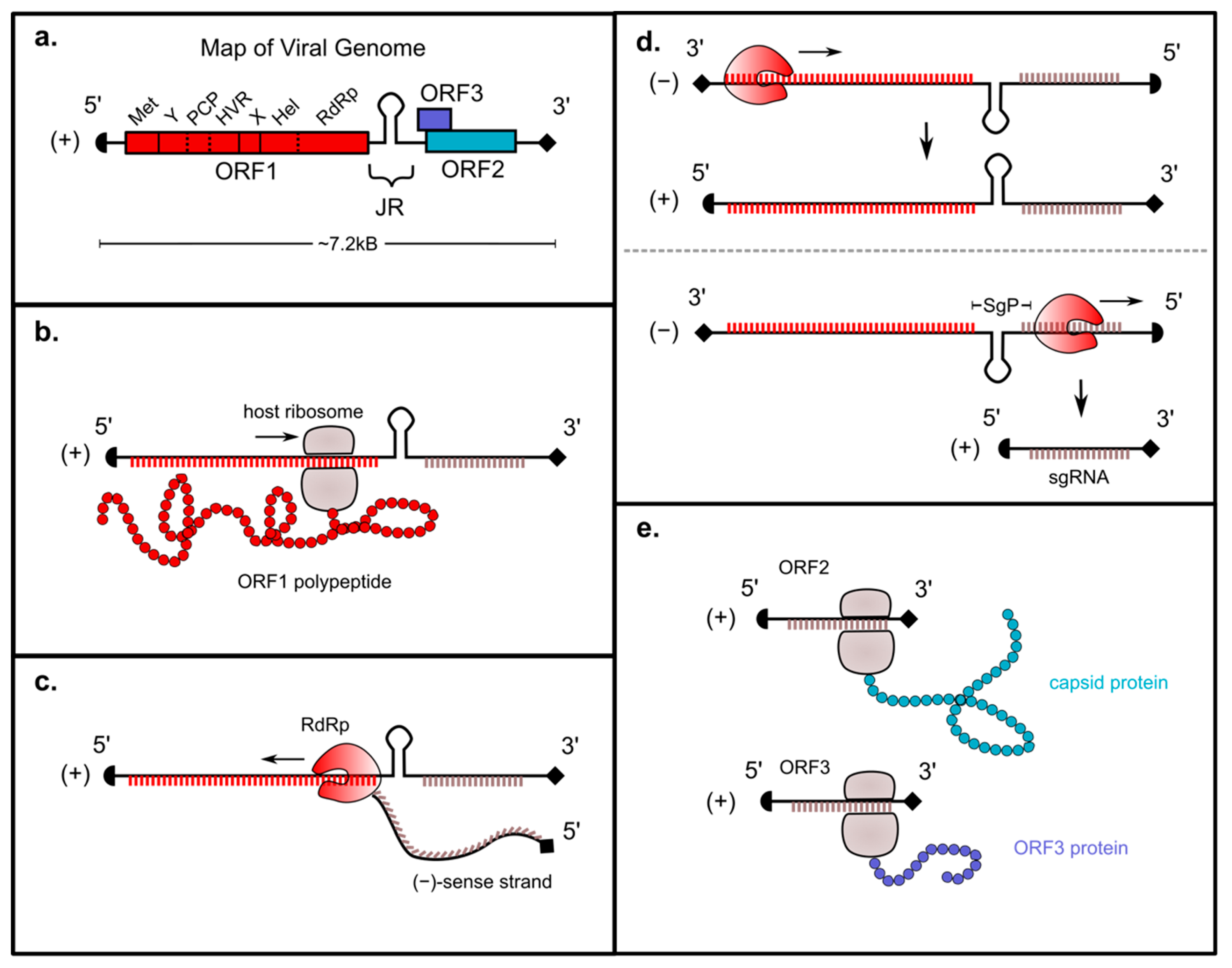
3. Genomic Replication Strategy
4. Roles of ORF1-Encoded Regions in HEV Replication
4.1. Methyltransferase
4.1.1. Introduction
4.1.2. Function
4.1.3. Clinical Relevance
4.2. X/Macro and Y Domains
4.2.1. Introduction
4.2.2. X Domain Functions
4.2.3. Y Domain Function
4.2.4. Clinical Relevance
4.3. Putative Protease Region
4.3.1. Introduction
4.3.2. Evidence that the Putative Protease Region is Responsible for Processing the HEV Polyprotein into Discrete Function Units
- Evidence for a cysteine protease that processes ORF1 into nine protein subunits: In silico analysis predicted a papain-like beta barrel fold and identified the proteolytic dyad of this putative papain-like cysteine protease (PCP) as residues C434 and H443 [41]. A putative zinc-binding motif and three potential disulfide bridges were also predicted in the PCP region [41]. To experimentally characterize this region, one group used a baculovirus expression system to overcome the low yield of ORF1 protein expressed in cells during natural infection. Using matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF), they observed nine distinct protein species of ORF1 of genotype 1 of HEV appearing in a time-dependent manner upon expression in non-natural host insect cells [42]. Additionally, they observed that treatment with a cysteine protease inhibitor blocked this processing of the ORF1 polyprotein [42].
- Evidence for processing of ORF1 into two subunits: Another study utilizing a GFP reporter replicon of the genotype 1 SAR55 strain of HEV showed that site-directed mutagenesis of the following residues in the PCP domain abrogated GFP expression in HUH 7 S10-3 cells, used in the study as a proxy for viral replication: C457A, C459A, C471A, C472A, C481A, C483A, H443L, H497L, and H590L [43]. This study also found that expressing histidine and HA-tagged replicons in HUH7 S10-3 cells showed the 186 kDa protein processed into approximately 35 and 78 kDa fragments.
- Evidence for chymotrypsin-like activity of the putative protease: One study found that purified and dialyzed fragments of HEV ORF1 and ORF2 were processed into smaller fragments when incubated with purified protein from the PCP region spanning amino acids 440–610, and that inhibitors to chymotrypsin halted this processing, suggesting that this region harbors a different class of proteolytic activity [44].
- Evidence for serine protease cleavage sites within the PCP region: Conversely, a recent study found two conserved putative cleavage sites for cellular thrombin and one putative cleavage site for cellular factor Xa within HEV ORF1, conserved across all HEV genotypes; the factor Xa site was suggested within the PCP domain at amino acid 560, and the thrombin sites in X domain (between 846 and 862 depending on HEV genotype) and in RNA-dependent RNA polymerase (between 1218 and 1235 depending on genotype) [45]. To characterize these sites, the authors made mutations in an HEV genotype 1 reporter replicon (GenBank accession no: AF444002.1) and found viral replication to be impeded, and that treating HUH7 S10-3 cells with a serine protease inhibitor or siRNA to interfere with these cellular factors also inhibited viral replication [45].
4.3.3. Evidence against Processing
4.4. Hypervariable Region
4.4.1. Introduction
4.4.2. Function
4.4.3. Insertions
4.5. Helicase
4.5.1. Introduction
4.5.2. Function
4.5.3. Clinical Relevance
4.6. RNA-Dependent RNA Polymerase
4.6.1. Introduction
4.6.2. Host and Viral Protein Interactions
4.6.3. Clinical Relevance and Therapeutic Potential
5. cis-Acting Regulatory Elements
6. Conclusions
- Which host factors are essential for HEV RNA replication?
- Does HEV replication lead to membrane rearrangements similar to other (+) RNA viruses?
- Does ORF1 polyprotein get processed? If so, how?
- Does the PCP region contain protease activity? If so, against what targets?
- What is the role of the HVR? How do insertions in this region confer cell culture adaptation?
- What is the role of the Y domain in the HEV replication cycle?
- What is the structure of the ORF1 polyprotein, or structures of ORF1-encoded proteins (if processed)?
- What are the kinetics and levels of transcription of antisense, full-length (+)-sense, and sgRNAs; and what are the mechanisms regulating expression of these elements?
- What are the components of the replicase complex for HEV? Does the RdRp self-oligomerize?
Funding
Acknowledgments
Conflicts of Interest
References
- Smith, D.B.; Simmonds, P.; Jameel, S.; Emerson, S.U.; Harrison, T.J.; Meng, X.J.; Okamoto, H.; Van Der Poel, W.H.M.; Purdy, M.A. Consensus proposals for classification of the family Hepeviridae. J. Gen. Virol. 2014, 95, 2223–2232. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Tanaka, T.; Takahashi, H.; Hoshino, Y.; Nagashima, S.; Mizuo, H.; Yazaki, Y.; Takagi, T.; Azuma, M.; Kusano, E.; et al. Hepatitis E Virus (HEV) Strains in Serum Samples Can Replicate Efficiently in Cultured Cells Despite the Coexistence of HEV Antibodies: Characterization of HEV Virions in Blood Circulation. J. Clin. Microbiol. 2010, 48, 1112–1125. [Google Scholar] [CrossRef] [PubMed]
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis E virus: Advances and challenges. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, M.S. Discovery of hepatitis E: The epidemic non-A, non-B hepatitis 30 years down the memory lane. Virus Res. 2011, 161, 3–14. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Lhomme, S.; Abravanel, F.; Marion, O.; Péron, J.M.; Alric, L.; Izopet, J. Treatment of HEV Infection in Patients with a Solid-Organ Transplant and Chronic Hepatitis. Viruses 2016, 8, 222. [Google Scholar] [CrossRef] [PubMed]
- Khuroo, M.; Kamili, S. Aetiology, clinical course and outcome of sporadic acute viral hepatitis in pregnancy. J. Viral Hepat. 2003, 10, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Mallet, V.; Izopet, J. Ribavirin for Chronic Hepatitis E Virus Infection. N. Engl. J. Med. 2014, 370, 2446–2448. [Google Scholar] [CrossRef] [PubMed]
- Borkakoti, J.; Ahmed, G.; Kar, P. Report of a novel C1483W mutation in the hepatitis E virus polymerase in patients with acute liver failure. Infect. Genet. Evol. 2016, 44, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A Mutation in the Hepatitis E Virus RNA Polymerase Promotes Its Replication and Associates with Ribavirin Treatment Failure in Organ Transplant Recipients. Gastroenterology 2014, 147, 1008–1011. [Google Scholar] [CrossRef]
- Debing, Y.; Ramière, C.; Dallmeier, K.; Piorkowski, G.; Trabaud, M.-A.; Lebossé, F.; Scholtès, C.; Roche, M.; Legras-Lachuer, C.; De Lamballerie, X.; et al. Hepatitis E virus mutations associated with ribavirin treatment failure result in altered viral fitness and ribavirin sensitivity. J. Hepatol. 2016, 65, 499–508. [Google Scholar] [CrossRef]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Shalimar; Nayak, B.; Ct, R.K.; Surjit, M. Endoplasmic Reticulum Stress Induced Synthesis of a Novel Viral Factor Mediates Efficient Replication of Genotype-1 Hepatitis E Virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Purcell, R.H.; Emerson, S.U. Identification of the 5′ terminal sequence of the SAR-55 and MEX-14 strains of hepatitis E virus and confirmation that the genome is capped. J. Med. Virol. 2001, 65, 293–295. [Google Scholar] [CrossRef] [PubMed]
- Varma, S.P.; Kumar, A.; Kapur, N.; Durgapal, H.; Acharya, S.K.; Panda, S.K. Hepatitis E virus replication involves alternating negative- and positive-sense RNA synthesis. J. Gen. Virol. 2011, 92, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Graff, J.; Torian, U.; Nguyen, H.; Emerson, S.U. A Bicistronic Subgenomic mRNA Encodes both the ORF2 and ORF3 Proteins of Hepatitis E Virus. J. Virol. 2006, 80, 5919–5926. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Heller, B.; Capuccino, J.M.V.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef] [PubMed]
- Jameel, S.; Zafrullah, M.; Ozdener, M.H.; Panda, S.K. Expression in animal cells and characterization of the hepatitis E virus structural proteins. J. Virol. 1996, 70, 207–216. [Google Scholar] [PubMed]
- Balayart, M.; Andjaparidze, A.; Savinskaya, S.; Ketiladze, E.; Braginsky, D.; Savinov, A.; Poleschük, V.; Balayan, M. Evidence for a Virus in Non-A, Non-B Hepatitis Transmitted via the Fecal-Oral Route. Intervirology 1983, 20, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Gorbalenya, A.E.; Purdy, M.A.; Rozanov, M.N.; Reyes, G.R.; Bradley, D.W. Computer-assisted assignment of functional domains in the nonstructural polyprotein of hepatitis E virus: Delineation of an additional group of positive-strand RNA plant and animal viruses. Proc. Natl. Acad. Sci. USA 1992, 89, 8259–8263. [Google Scholar] [CrossRef] [PubMed]
- Kabrane-Lazizi, Y.; Meng, X.J.; Purcell, R.H.; Emerson, S.U. Evidence that the Genomic RNA of Hepatitis E Virus Is Capped. J. Virol. 1999, 73, 8848–8850. [Google Scholar]
- Magden, J.; Takeda, N.; Li, T.; Auvinen, P.; Ahola, T.; Miyamura, T.; Merits, A.; Kääriäinen, L. Virus-specific mRNA capping enzyme encoded by hepatitis E virus. J. Virol. 2001, 75, 6249–6255. [Google Scholar] [CrossRef]
- Ahola, T.; Laakkonen, P.; Vihinen, H.; Kääriäinen, L. Critical residues of Semliki Forest virus RNA capping enzyme involved in methyltransferase and guanylyltransferase-like activities. J. Virol. 1997, 71, 392–397. [Google Scholar] [PubMed]
- Laakkonen, P.; Hyvönen, M.; Peränen, J.; Kääriäinen, L. Expression of Semliki Forest virus nsP1-specific methyltransferase in insect cells and in Escherichia coli. J. Virol. 1994, 68, 7418–7425. [Google Scholar] [PubMed]
- Mi, S.; Durbin, R.; Huang, H.V.; Rice, C.M.; Stollar, V. Association of the sindbis virus RNA methyltransferase activity with the nonstructural protein nsP1. Virology 1989, 170, 385–391. [Google Scholar] [CrossRef]
- Rozanov, M.N.; Koonin, E.V.; Gorbalenya, A.E. Conservation of the putative methyltransferase domain: A hallmark of the ‘Sindbis-like’ supergroup of positive-strand RNA viruses. J. Gen. Virol. 1992, 73, 2129–2134. [Google Scholar] [CrossRef] [PubMed]
- Scheidel, L.M.; Durbin, R.K.; Stollar, V. SVLM21, a Sindbis virus mutant resistant to methionine deprivation, encodes an altered methyltransferase. Virology 1989, 173, 408–414. [Google Scholar] [CrossRef]
- Ahola, T.; Kääriäinen, L. Reaction in alphavirus mRNA capping: Formation of a covalent complex of nonstructural protein nsP1 with 7-methyl-GMP. Proc. Natl. Acad. Sci. USA 1995, 92, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Boon, J.A.D.; Ahlquist, P. Helicase and Capping Enzyme Active Site Mutations in Brome Mosaic Virus Protein 1a Cause Defects in Template Recruitment, Negative-Strand RNA Synthesis, and Viral RNA Capping. J. Virol. 2000, 74, 8803–8811. [Google Scholar] [CrossRef] [PubMed]
- Borkakoti, J.; Ahmed, G.; Rai, A.; Kar, P. Report of novel H105R, D29N, V27A mutations in the methyltransferase region of the HEV genome in patients with acute liver failure. J. Clin. Virol. 2017, 91, 1–4. [Google Scholar] [CrossRef]
- Mishra, N.; Walimbe, A.M.; Arankalle, V.A. Hepatitis E virus from India exhibits significant amino acid mutations in fulminant hepatic failure patients. Virus Genes 2013, 46, 47–53. [Google Scholar] [CrossRef]
- Karras, I.G.; Kustatscher, G.; Buhecha, H.R.; Allen, M.D.; Pugieux, C.; Sait, F.; Bycroft, M.; Ladurner, A.G. The macro domain is an ADP-ribose binding module. EMBO J. 2005, 24, 1911–1920. [Google Scholar] [CrossRef]
- Amé, J.C.; Spenlehauer, C.; De Murcia, G.; Amé, J. The PARP superfamily. BioEssays 2004, 26, 882–893. [Google Scholar] [CrossRef] [PubMed]
- Corda, D.; Di Girolamo, M. Functional aspects of protein mono-ADP-ribosylation. EMBO J. 2003, 22, 1953–1958. [Google Scholar] [CrossRef] [PubMed]
- Neuvonen, M.; Ahola, T. Differential Activities of Cellular and Viral Macro Domain Proteins in Binding of ADP-Ribose Metabolites. J. Mol. Biol. 2009, 385, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Parvez, M.K. The hepatitis E virus ORF1 ‘X-domain’ residues form a putative macrodomain protein/Appr-1″-pase catalytic-site, critical for viral RNA replication. Gene 2015, 566, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Anang, S.; Subramani, C.; Nair, V.P.; Kaul, S.; Kaushik, N.; Sharma, C.; Tiwari, A.; Ranjith-Kumar, C.; Surjit, M. Identification of critical residues in Hepatitis E virus macro domain involved in its interaction with viral methyltransferase and ORF3 proteins. Sci. Rep. 2016, 6, 25133. [Google Scholar] [CrossRef] [PubMed]
- Parvez, M.K. Mutational analysis of hepatitis E virus ORF1 “Y-domain”: Effects on RNA replication and virion infectivity. World J. Gastroenterol. 2017, 23, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Ahola, T.; Karlin, D.G. Sequence analysis reveals a conserved extension in the capping enzyme of the alphavirus supergroup, and a homologous domain in nodaviruses. Biol. Direct 2015, 10, 197. [Google Scholar] [CrossRef]
- Lhomme, S.; Garrouste, C.; Kamar, N.; Saune, K.; Abravanel, F.; Mansuy, J.M.; Dubois, M.; Rostaing, L.; Izopet, J. Influence of polyproline region and macro domain genetic heterogeneity on HEV persistence in immunocompromised patients. J. Infect. Dis. 2014, 209, 300–303. [Google Scholar] [CrossRef]
- Ojha, N.K.; Lole, K.S. Hepatitis E virus ORF1 encoded macro domain protein interacts with light chain subunit of human ferritin and inhibits its secretion. Mol. Cell. Biochem. 2016, 417, 75–85. [Google Scholar] [CrossRef]
- Eriksson, K.K.; Cervantes-Barragán, L.; Ludewig, B.; Thiel, V. Mouse Hepatitis Virus Liver Pathology Is Dependent on ADP-Ribose-1″-Phosphatase, a Viral Function Conserved in the Alpha-Like Supergroup. J. Virol. 2008, 82, 12325–12334. [Google Scholar] [CrossRef]
- Parvez, M.K.; Khan, A.A. Molecular modeling and analysis of hepatitis E virus (HEV) papain-like cysteine protease. Virus Res. 2014, 179, 220–224. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, D.; Thomas, S.; Chakraborty, M.; Jameel, S. Expression and processing of the Hepatitis E virus ORF1 nonstructural polyprotein. Virol. J. 2006, 3, 38. [Google Scholar] [CrossRef] [PubMed]
- Parvez, M.K. Molecular characterization of hepatitis E virus ORF1 gene supports a papain-like cysteine protease (PCP)-domain activity. Virus Res. 2013, 178, 553–556. [Google Scholar] [CrossRef] [PubMed]
- Paliwal, D.; Panda, S.K.; Kapur, N.; Varma, S.P.K.; Durgapal, H. Hepatitis E virus (HEV) protease: A chymotrypsin-like enzyme that processes both non-structural (pORF1) and capsid (pORF2) protein. J. Gen. Virol. 2014, 95, 1689–1700. [Google Scholar] [CrossRef] [PubMed]
- Kanade, G.D.; Pingale, K.D.; Karpe, Y.A. Activities of Thrombin and Factor Xa Are Essential for Replication of Hepatitis E Virus and Are Possibly Implicated in ORF1 Polyprotein Processing. J. Virol. 2018, 92, e01853-17. [Google Scholar] [CrossRef] [PubMed]
- Perttilä, J.; Spuul, P.; Ahola, T. Early secretory pathway localization and lack of processing for hepatitis E virus replication protein pORF1. J. Gen. Virol. 2013, 94, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, S.; Zhou, Y.; Frey, T.K. Lack of Processing of the Expressed ORF1 Gene Product of Hepatitis E Virus. Virol. J. 2011, 8, 245. [Google Scholar] [CrossRef] [PubMed]
- Ansari, I.H.; Nanda, S.K.; Durgapal, H.; Agrawal, S.; Mohanty, S.K.; Gupta, D.; Jameel, S.; Panda, S.K. Cloning, sequencing, and expression of the hepatitis E virus (HEV) nonstructural open reading frame 1 (ORF1). J. Med. Virol. 2000, 60, 275–283. [Google Scholar] [CrossRef]
- Ropp, S.L.; Tam, A.W.; Beames, B.; Purdy, M.; Frey, T.K. Expression of the hepatitis E virus ORF1. Arch. Virol. 2000, 145, 1321–1337. [Google Scholar] [CrossRef]
- Karpe, Y.A.; Lole, K.S. Deubiquitination activity associated with hepatitis E virus putative papain-like cysteine protease. J. Gen. Virol. 2011, 92, 2088–2092. [Google Scholar] [CrossRef]
- Purdy, M.A.; Lara, J.; Khudyakov, Y.E. The Hepatitis E Virus Polyproline Region Is Involved in Viral Adaptation. PLoS ONE 2012, 7, e35974. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Vanek, J.; Ramalingam, S.; Johannessen, I.; Templeton, K.; Simmonds, P. Evolution of the hepatitis E virus hypervariable region. J. Gen. Virol. 2012, 93, 2408–2418. [Google Scholar] [CrossRef] [PubMed]
- Pudupakam, R.S.; Huang, Y.W.; Opriessnig, T.; Halbur, P.G.; Pierson, F.W.; Meng, X.J. Deletions of the hypervariable region (HVR) in open reading frame 1 of hepatitis E virus do not abolish virus infectivity: Evidence for attenuation of HVR deletion mutants in vivo. J. Virol. 2009, 83, 384–395. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus–host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef] [PubMed]
- Pudupakam, R.S.; Kenney, S.P.; Córdoba, L.; Huang, Y.W.; Dryman, B.A.; Leroith, T.; Pierson, F.W.; Meng, X.J. Mutational Analysis of the Hypervariable Region of Hepatitis E Virus Reveals Its Involvement in the Efficiency of Viral RNA Replication. J. Virol. 2011, 85, 10031–10040. [Google Scholar] [CrossRef] [PubMed]
- Lhomme, S.; Abravanel, F.; Dubois, M.; Sandres-Saune, K.; Mansuy, J.-M.; Rostaing, L.; Kamar, N.; Izopet, J. Characterization of the Polyproline Region of the Hepatitis E Virus in Immunocompromised Patients. J. Virol. 2014, 88, 12017–12025. [Google Scholar] [CrossRef] [PubMed]
- Johne, R.; Reetz, J.; Ulrich, R.G.; Machnowska, P.; Sachsenröder, J.; Nickel, P.; Hofmann, J. An ORF1-rearranged hepatitis E virus derived from a chronically infected patient efficiently replicates in cell culture. J. Viral. Hepat. 2014, 21, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Torian, U.; Faulk, K.; Mather, K.; Engle, R.E.; Thompson, E.; Bonkovsky, H.L.; Emerson, S.U. A naturally occurring human/hepatitis E recombinant virus predominates in serum but not in faeces of a chronic hepatitis E patient and has a growth advantage in cell culture. J. Gen. Virol. 2012, 93, 526–530. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Nguyen, H.T.; Faulk, K.; Mather, K.; Torian, U.; Engle, R.E.; Emerson, S.U. Adaptation of a Genotype 3 Hepatitis E Virus to Efficient Growth in Cell Culture Depends on an Inserted Human Gene Segment Acquired by Recombination. J. Virol. 2012, 86, 5697–5707. [Google Scholar] [CrossRef]
- Kenney, S.P.; Meng, X.J. The Lysine Residues within the Human Ribosomal Protein S17 Sequence Naturally Inserted into the Viral Nonstructural Protein of a Unique Strain of Hepatitis E Virus Are Important for Enhanced Virus Replication. J. Virol. 2015, 89, 3793–3803. [Google Scholar] [CrossRef]
- Kadaré, G.; Haenni, A.L. Virus-encoded RNA helicases. J. Virol. 1997, 71, 2583–2590. [Google Scholar]
- Mhaindarkar, V.; Sharma, K.; Lole, K.S. Mutagenesis of hepatitis E virus helicase motifs: Effects on enzyme activity. Virus Res. 2014, 179, 26–33. [Google Scholar] [CrossRef]
- Karpe, Y.A.; Lole, K.S. RNA 5′-Triphosphatase Activity of the Hepatitis E Virus Helicase Domain. J. Virol. 2010, 84, 9637–9641. [Google Scholar] [CrossRef]
- Karpe, Y.A.; Lole, K.S. NTPase and 5′ to 3′ RNA Duplex-Unwinding Activities of the Hepatitis E Virus Helicase Domain. J. Virol. 2010, 84, 3595–3602. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.; Ni, Y.Y.; Meng, X.J. Substitution of amino acid residue V1213 in the helicase domain of the genotype 3 hepatitis E virus reduces virus replication. Virol. J. 2018, 15, 32. [Google Scholar] [CrossRef] [PubMed]
- Devhare, P.; Sharma, K.; Mhaindarkar, V.; Arankalle, V.; Lole, K. Analysis of helicase domain mutations in the hepatitis E virus derived from patients with fulminant hepatic failure: Effects on enzymatic activities and virus replication. Virus Res. 2014, 184, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Parvez, M.K.; Subbarao, N. Molecular Analysis and Modeling of Hepatitis E Virus Helicase and Identification of Novel Inhibitors by Virtual Screening. BioMed Res. Int. 2018, 2018, 1–8. [Google Scholar] [CrossRef]
- Koonin, E.V. The phylogeny of RNA-dependent RNA polymerases of positive-strand RNA viruses. J. Gen. Virol. 1991, 72, 2197–2206. [Google Scholar] [CrossRef]
- Van Der Heijden, M.W.; Bol, J.F. Composition of alphavirus-like replication complexes: Involvement of virus and host encoded proteins. Arch. Virol. 2002, 147, 875–898. [Google Scholar] [CrossRef]
- Ichiyama, K.; Yamada, K.; Tanaka, T.; Nagashima, S.; Jirintai; Takahashi, M.; Okamoto, H. Determination of the 5′-terminal sequence of subgenomic RNA of hepatitis E virus strains in cultured cells. Arch. Virol. 2009, 154, 1945–1951. [Google Scholar] [CrossRef]
- Cao, D.; Huang, Y.W.; Meng, X.J. The Nucleotides on the Stem-Loop RNA Structure in the Junction Region of the Hepatitis E Virus Genome Are Critical for Virus Replication. J. Virol. 2010, 84, 13040–13044. [Google Scholar] [CrossRef] [PubMed]
- Mahilkar, S.; Paingankar, M.S.; Lole, K.S. Hepatitis E virus RNA dependent RNA polymerase: RNA template specificities, recruitment and synthesis. J. Gen. Virol. 2016, 97, 2231–2242. [Google Scholar] [CrossRef] [PubMed]
- Osterman, A.; Stellberger, T.; Gebhardt, A.; Kurz, M.; Friedel, C.C.; Uetz, P.; Nitschko, H.; Baiker, A.; Vizoso-Pinto, M.G. The Hepatitis E virus intraviral interactome. Sci. Rep. 2015, 5, 13872. [Google Scholar] [CrossRef]
- Hobson, S.D.; Rosenblum, E.S.; Richards, O.C.; Richmond, K.; Kirkegaard, K.; Schultz, S.C. Oligomeric structures of poliovirus polymerase are important for function. EMBO J. 2001, 20, 1153–1163. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.M.; Hockman, M.A.; Staschke, K.; Johnson, R.B.; Case, K.A.; Lu, J.; Parsons, S.; Zhang, F.; Rathnachalam, R.; Kirkegaard, K.; et al. Oligomerization and Cooperative RNA Synthesis Activity of Hepatitis C Virus RNA-Dependent RNA Polymerase. J. Virol. 2002, 76, 3865–3872. [Google Scholar] [CrossRef][Green Version]
- Pingale, K.D.; Kanade, G.D.; Karpe, Y.A. Hepatitis E virus polymerase binds to IFIT1 to protect the viral RNA from IFIT1-mediated translation inhibition. J. Gen. Virol. 2019, 100, 471–483. [Google Scholar] [CrossRef] [PubMed]
- Haldipur, B.; Bhukya, P.L.; Arankalle, V.; Lole, K. Positive Regulation of Hepatitis E Virus Replication by MicroRNA-122. J. Virol. 2018, 92, e01999-17. [Google Scholar] [CrossRef]
- Jopling, C.L.; Yi, M.; Lancaster, A.M.; Lemon, S.M.; Sarnow, P. Modulation of Hepatitis C Virus RNA Abundance by a Liver-Specific MicroRNA. Science 2005, 309, 1577–1581. [Google Scholar] [CrossRef]
- Kaur, M.; Hyams, K.C.; Purdy, M.A.; Krawczyński, K.; Ching, W.M.; Fry, K.E.; Reyes, G.R.; Bradley, D.W.; Carl, M. Human linear B-cell epitopes encoded by the hepatitis E virus include determinants in the RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. USA 1992, 89, 3855–3858. [Google Scholar] [CrossRef]
- Todt, D.; Gisa, A.; Radonic, A.; Nitsche, A.; Behrendt, P.; Suneetha, P.V.; Pischke, S.; Bremer, B.; Brown, R.J.P.; Manns, M.P.; et al. In vivo evidence for ribavirin-induced mutagenesis of the hepatitis E virus genome. Gut 2016, 65, 1733–1743. [Google Scholar] [CrossRef]
- Kamar, N.; Selves, J.; Mansuy, J.M.; Ouezzani, L.; Péron, J.M.; Guitard, J.; Cointault, O.; Esposito, L.; Abravanel, F.; Danjoux, M.; et al. Hepatitis E Virus and Chronic Hepatitis in Organ-Transplant Recipients. N. Engl. J. Med. 2008, 358, 811–817. [Google Scholar] [CrossRef] [PubMed]
- Kaushik, N.; Subramani, C.; Anang, S.; Muthumohan, R.; Shalimar; Nayak, B.; Ranjith-Kumar, C.T.; Surjit, M. Zinc Salts Block Hepatitis E Virus Replication by Inhibiting the Activity of Viral RNA-Dependent RNA Polymerase. J. Virol. 2017, 91, e00754-17. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Hua, X.; Yang, S.; Yuan, C.; Zhang, W. Effective inhibition of hepatitis E virus replication in A549 cells and piglets by RNA interference (RNAi) targeting RNA-dependent RNA polymerase. Antivir. Res. 2009, 83, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Agrawal, S.; Gupta, D.; Panda, S.K. The 3′ End of Hepatitis E Virus (HEV) Genome Binds Specifically to the Viral RNA-Dependent RNA Polymerase (RdRp). Virology 2001, 282, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Niesters, H.G.; Strauss, J.H. Mutagenesis of the conserved 51-nucleotide region of Sindbis virus. J. Virol. 1990, 64, 1639–1647. [Google Scholar] [PubMed]
- Grakoui, A.; Levis, R.; Raju, R.; Huang, H.V.; Rice, C.M. A cis-acting mutation in the Sindbis virus junction region which affects subgenomic RNA synthesis. J. Virol. 1989, 63, 5216–5227. [Google Scholar] [PubMed]
- Graff, J.; Nguyen, H.; Kasorndorkbua, C.; Halbur, P.G.; St Claire, M.; Purcell, R.H.; Emerson, S.U. In vitro and in vivo mutational analysis of the 3′-terminal regions of hepatitis e virus genomes and replicons. J. Virol. 2005, 79, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Nimgaonkar, I.; Archer, N.F.; Bram, Y.; Heller, B.; Schwartz, R.E.; Ploss, A. Identification of the Intragenomic Promoter Controlling Hepatitis E Virus Subgenomic RNA Transcription. mBio 2018, 9, e00769-18. [Google Scholar] [CrossRef]
- Cao, D.; Ni, Y.Y.; Walker, M.; Huang, Y.W.; Meng, X.J. Roles of the genomic sequence surrounding the stem-loop structure in the junction region including the 3′ terminus of open reading frame 1 in hepatitis E virus replication. J. Med. Virol. 2018, 90, 1524–1531. [Google Scholar] [CrossRef]
- Huang, Y.W.; Opriessnig, T.; Halbur, P.G.; Meng, X.J. Initiation at the Third In-Frame AUG Codon of Open Reading Frame 3 of the Hepatitis E Virus Is Essential for Viral Infectivity In Vivo. J. Virol. 2007, 81, 3018–3026. [Google Scholar] [CrossRef]
- Tam, A.W.; Smith, M.M.; Guerra, M.E.; Huang, C.C.; Bradley, D.W.; Fry, K.E.; Reyes, G.R. Hepatitis E virus (HEV): Molecular cloning and sequencing of the full-length viral genome. Virology 1991, 185, 120–131. [Google Scholar] [CrossRef]

© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
LeDesma, R.; Nimgaonkar, I.; Ploss, A. Hepatitis E Virus Replication. Viruses 2019, 11, 719. https://doi.org/10.3390/v11080719
LeDesma R, Nimgaonkar I, Ploss A. Hepatitis E Virus Replication. Viruses. 2019; 11(8):719. https://doi.org/10.3390/v11080719
Chicago/Turabian StyleLeDesma, Robert, Ila Nimgaonkar, and Alexander Ploss. 2019. "Hepatitis E Virus Replication" Viruses 11, no. 8: 719. https://doi.org/10.3390/v11080719
APA StyleLeDesma, R., Nimgaonkar, I., & Ploss, A. (2019). Hepatitis E Virus Replication. Viruses, 11(8), 719. https://doi.org/10.3390/v11080719