Hepatitis E Virus Entry



{kind=link}
{kind=link}
Abstract
1. Introduction
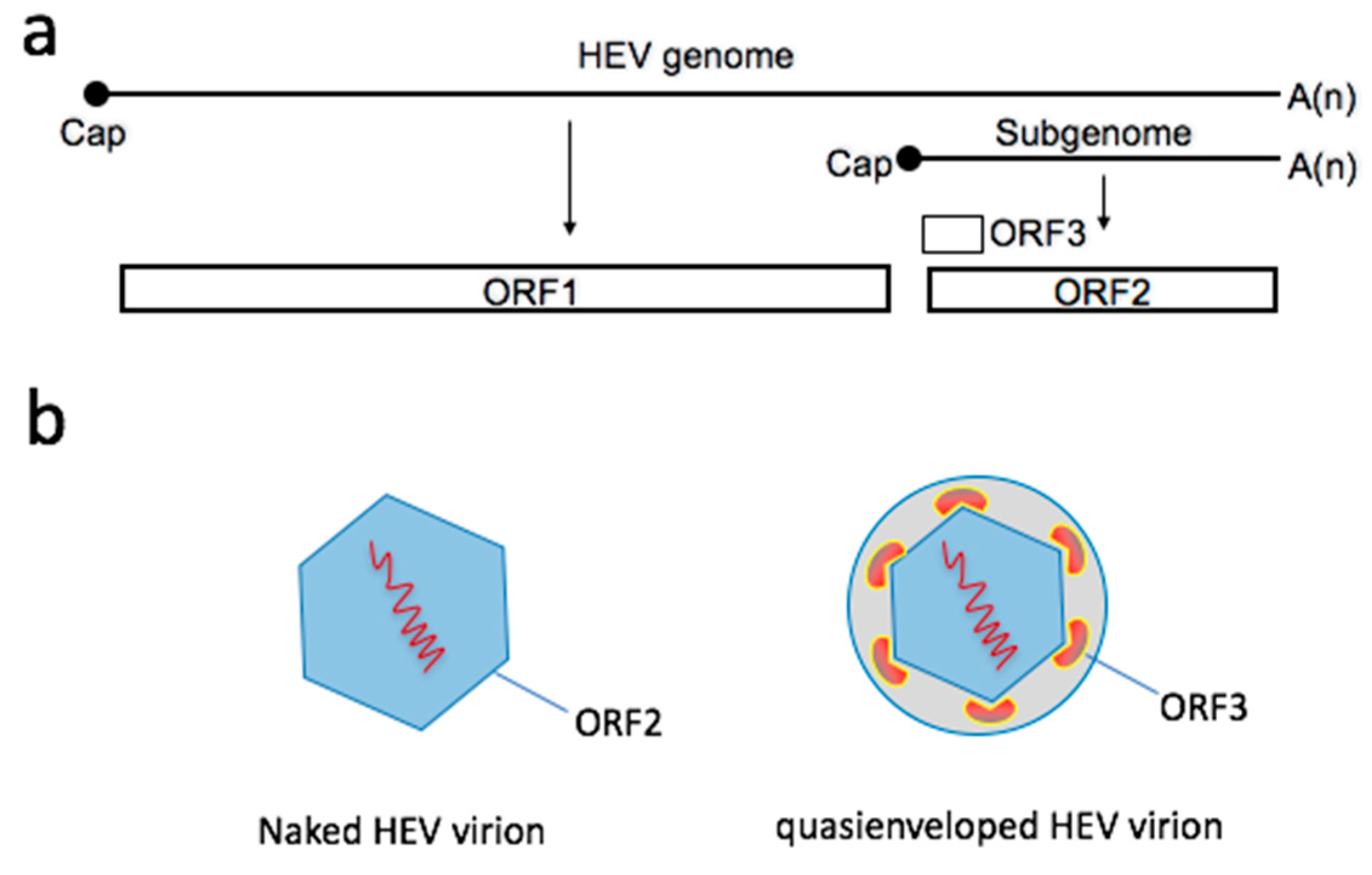
2. Two Types of Virions Naturally Exist during an HEV Infection
3. Cell Entry of Naked HEV
3.1. Cell Attachment
3.1.1. HSGPs (Heparan Sulfate Proteoglycans)
3.1.2. GRP78 (Glucose-Regulated Protein 78)
3.1.3. ASGPR (Asialoglycoprotein Receptor)
3.1.4. ATP5B (ATP Synthase Subunit 5β)
3.1.5. ITGA3 (Integrin Alpha 3)
3.2. Internalization and Intracellular Trafficking
3.3. Uncoating
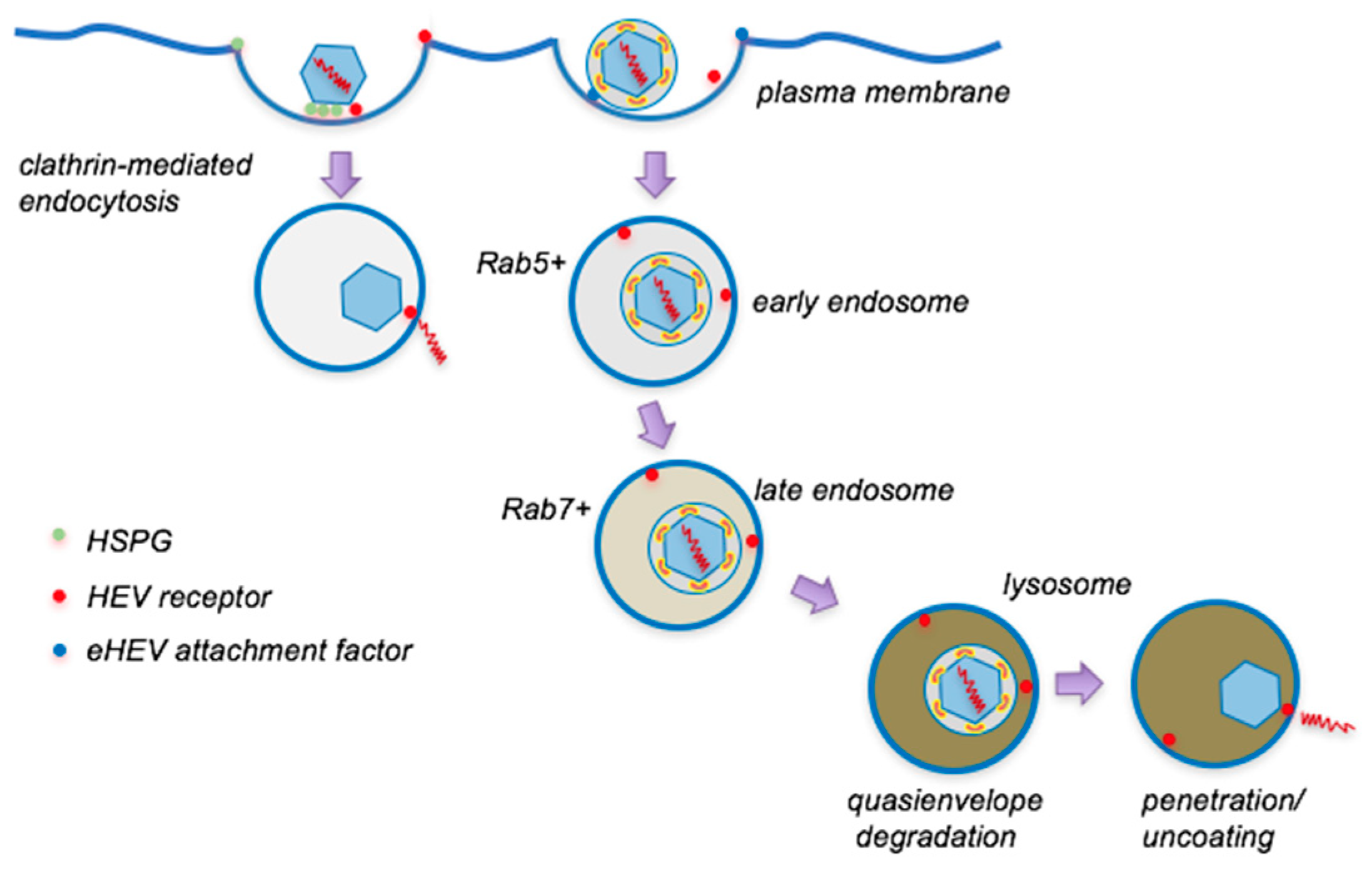
4. Entry of Quasienveloped HEV
4.1. Cell Attachment
4.2. Internalization and Intracellular Trafficking
4.3. Uncoating
5. Summary
Funding
Conflicts of Interest
References
- Nimgaonkar, I.; Ding, Q.; Schwartz, R.E.; Ploss, A. Hepatitis E virus: Advances and challenges. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 96–110. [Google Scholar] [CrossRef] [PubMed]
- Kenney, S.P. The Current Host Range of Hepatitis E Viruses. Viruses 2019, 11, 452. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Rostaing, L.; Abravanel, F.; Garrouste, C.; Lhomme, S.; Esposito, L.; Basse, G.; Cointault, O.; Ribes, D.; Nogier, M.B.; et al. Ribavirin therapy inhibits viral replication on patients with chronic hepatitis e virus infection. Gastroenterology 2010, 139, 1612–1618. [Google Scholar] [CrossRef] [PubMed]
- Kamar, N.; Izopet, J.; Tripon, S.; Bismuth, M.; Hillaire, S.; Dumortier, J.; Radenne, S.; Coilly, A.; Garrigue, V.; D’Alteroche, L.; et al. Ribavirin for chronic hepatitis E virus infection in transplant recipients. N. Engl. J. Med. 2014, 370, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Gisa, A.; Dallmeier, K.; Pischke, S.; Bremer, B.; Manns, M.; Wedemeyer, H.; Suneetha, P.V.; Neyts, J. A mutation in the hepatitis E virus RNA polymerase promotes its replication and associates with ribavirin treatment failure in organ transplant recipients. Gastroenterology 2014, 147, 1008–1011.e7, quiz e15-6. [Google Scholar] [CrossRef] [PubMed]
- Debing, Y.; Ramiere, C.; Dallmeier, K.; Piorkowski, G.; Trabaud, M.A.; Lebosse, F.; Scholtes, C.; Roche, M.; Legras-Lachuer, C.; de Lamballerie, X.; et al. Hepatitis E virus mutations associated with ribavirin treatment failure result in altered viral fitness and ribavirin sensitivity. J. Hepatol. 2016, 65, 499–508. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.C.; Zhang, J.; Zhang, X.F.; Zhou, C.; Wang, Z.Z.; Huang, S.J.; Wang, H.; Yang, C.L.; Jiang, H.M.; Cai, J.P.; et al. Efficacy and safety of a recombinant hepatitis E vaccine in healthy adults: A large-scale, randomised, double-blind placebo-controlled, phase 3 trial. Lancet 2010, 376, 895–902. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, X.-F.; Huang, S.-J.; Wu, T.; Hu, Y.-M.; Wang, Z.-Z.; Wang, H.; Jiang, H.-M.; Wang, Y.-J.; Yan, Q.; et al. Long-Term Efficacy of a Hepatitis E Vaccine. N. Engl. J. Med. 2015, 372, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Balayan, M.S.; Andjaparidze, A.G.; Savinskaya, S.S.; Ketiladze, E.S.; Braginsky, D.M.; Savinov, A.P.; Poleschuk, V.F. Evidence for a virus in non-A, non-B hepatitis transmitted via the fecal-oral route. Intervirology 1983, 20, 23–31. [Google Scholar] [PubMed]
- Takahashi, M.; Tanaka, T.; Takahashi, H.; Hoshino, Y.; Nagashima, S.; Jirintai; Mizuo, H.; Yazaki, Y.; Takagi, T.; Azuma, M.; et al. Hepatitis E Virus (HEV) strains in serum samples can replicate efficiently in cultured cells despite the coexistence of HEV antibodies: Characterization of HEV virions in blood circulation. J. Clin. Microbiol. 2010, 48, 1112–1125. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Kobayashi, T.; Nishizawa, T.; Nishiyama, T.; Primadharsini, P.P.; Okamoto, H. Characterization of the Quasi-Enveloped Hepatitis E Virus Particles Released by the Cellular Exosomal Pathway. J. Virol. 2017, 91, e00822-17. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Li, X.; Feng, Z. Role of Envelopment in the HEV Life Cycle. Viruses 2016, 8, 229. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Ambardekar, C.; Lu, Y.; Feng, Z. Distinct Entry Mechanisms for Nonenveloped and Quasi-Enveloped Hepatitis E Viruses. J. Virol. 2016, 90, 4232–4242. [Google Scholar] [CrossRef] [PubMed]
- Tam, A.W.; Smith, M.M.; Guerra, M.E.; Huang, C.C.; Bradley, D.W.; Fry, K.E.; Reyes, G.R. Hepatitis E virus (HEV): Molecular cloning and sequencing of the full-length viral genome. Virology 1991, 185, 120–131. [Google Scholar] [CrossRef]
- Yamada, K.; Takahashi, M.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Nagashima, S.; Tanaka, T.; Okamoto, H. ORF3 protein of hepatitis E virus is essential for virion release from infected cells. J. Gen. Virol. 2009, 90, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.P.; Anang, S.; Subramani, C.; Madhvi, A.; Bakshi, K.; Srivastava, A.; Shalimar; Nayak, B.; Ranjith Kumar, C.T.; Surjit, M. Endoplasmic Reticulum Stress Induced Synthesis of a Novel Viral Factor Mediates Efficient Replication of Genotype-1 Hepatitis E Virus. PLoS Pathog. 2016, 12, e1005521. [Google Scholar] [CrossRef]
- Yamashita, T.; Mori, Y.; Miyazaki, N.; Cheng, R.H.; Yoshimura, M.; Unno, H.; Shima, R.; Moriishi, K.; Tsukihara, T.; Li, T.C.; et al. Biological and immunological characteristics of hepatitis E virus-like particles based on the crystal structure. Proc. Natl. Acad. Sci. USA 2009, 106, 12986–12991. [Google Scholar] [CrossRef] [PubMed]
- Xing, L.; Li, T.C.; Mayazaki, N.; Simon, M.N.; Wall, J.S.; Moore, M.; Wang, C.Y.; Takeda, N.; Wakita, T.; Miyamura, T.; et al. Structure of hepatitis E virion-sized particle reveals an RNA-dependent viral assembly pathway. J. Biol. Chem. 2010, 285, 33175–33183. [Google Scholar] [CrossRef]
- Guu, T.S.Y.; Liu, Z.; Ye, Q.; Mata, D.A.; Li, K.; Yin, C.; Zhang, J.; Tao, Y.J. Structure of the hepatitis E virus-like particle suggests mechanisms for virus assembly and receptor binding. Proc. Natl. Acad. Sci. USA 2009, 106, 12992–12997. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.J. Recent advances in Hepatitis E virus. J. Viral Hepat. 2010, 17, 153–161. [Google Scholar] [CrossRef]
- Li, S.; Tang, X.; Seetharaman, J.; Yang, C.; Gu, Y.; Zhang, J.; Du, H.; Shih, J.W.; Hew, C.L.; Sivaraman, J.; et al. Dimerization of hepatitis E virus capsid protein E2s domain is essential for virus-host interaction. PLoS Pathog. 2009, 5, e1000537. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Hirai-Yuki, A.; McKnight, K.L.; Lemon, S.M. Naked viruses that are’t always naked: Quasi-enveloped agents of acute hepatitis. Annu. Rev. Virol. 2014, 1, 539–560. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Yamada, K.; Hoshino, Y.; Takahashi, H.; Ichiyama, K.; Tanaka, T.; Okamoto, H. Monoclonal antibodies raised against the ORF3 protein of hepatitis E virus (HEV) can capture HEV particles in culture supernatant and serum but not those in feces. Arch. Virol. 2008, 153, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Lemon, S.M. Peek-a-boo: Membrane hijacking and the pathogenesis of viral hepatitis. Trends Microbiol. 2014, 22, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Ju, X.; Ding, Q. Hepatitis E Virus Assembly and Release. Viruses 2019, 11, 539. [Google Scholar] [CrossRef] [PubMed]
- Ren, X.; Hurley, J.H. Proline-rich regions and motifs in trafficking: From ESCRT interaction to viral exploitation. Traffic 2011, 12, 1282–1290. [Google Scholar] [CrossRef]
- Nagashima, S.; Takahashi, M.; Jirintai; Tanaka, T.; Yamada, K.; Nishizawa, T.; Okamoto, H. A PSAP motif in the ORF3 protein of hepatitis E virus is necessary for virion release from infected cells. J. Gen. Virol. 2011, 92, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Nagashima, S.; Jirintai, S.; Takahashi, M.; Kobayashi, T.; Tanggis; Nishizawa, T.; Kouki, T.; Yashiro, T.; Okamoto, H. Hepatitis E virus egress depends on the exosomal pathway, with secretory exosomes derived from multivesicular bodies. J. Gen. Virol. 2014, 95, 2166–2175. [Google Scholar] [CrossRef]
- McKnight, K.L.; Xie, L.; Gonzalez-Lopez, O.; Rivera-Serrano, E.E.; Chen, X.; Lemon, S.M. Protein composition of the hepatitis A virus quasi-envelope. Proc. Natl. Acad. Sci. USA 2017, 114, 6587–6592. [Google Scholar] [CrossRef]
- Van Cuyck-Gandre, H.; Cockman-Thomas, R.; Caudill, J.D.; Asher, L.S.; Armstrong, K.L.; Hauroeder, B.; Clements, N.J.; Binn, L.N.; Longer, C.F. Experimental African HEV infection in cynomolgus macaques (Macaca fascicularis). J. Med. Virol. 1998, 55, 197–202. [Google Scholar] [CrossRef]
- Longer, C.F.; Denny, S.L.; Caudill, J.D.; Miele, T.A.; Asher, L.V.; Myint, K.S.; Huang, C.C.; Engler, W.F.; LeDuc, J.W.; Binn, L.N.; et al. Experimental hepatitis E: Pathogenesis in cynomolgus macaques (Macaca fascicularis). J. Infect. Dis. 1993, 168, 602–609. [Google Scholar] [CrossRef] [PubMed]
- Wilen, C.B.; Lee, S.; Hsieh, L.L.; Orchard, R.C.; Desai, C.; Hykes, B.L., Jr.; McAllaster, M.R.; Balce, D.R.; Feehley, T.; Brestoff, J.R.; et al. Tropism for tuft cells determines immune promotion of norovirus pathogenesis. Science 2018, 360, 204–208. [Google Scholar] [CrossRef] [PubMed]
- Drave, S.A.; Debing, Y.; Walter, S.; Todt, D.; Engelmann, M.; Friesland, M.; Wedemeyer, H.; Neyts, J.; Behrendt, P.; Steinmann, E. Extra-hepatic replication and infection of hepatitis E virus in neuronal-derived cells. J. Viral Hepat. 2016, 23, 512–521. [Google Scholar] [CrossRef] [PubMed]
- Knegendorf, L.; Drave, S.A.; Dao Thi, V.L.; Debing, Y.; Brown, R.J.P.; Vondran, F.W.R.; Resner, K.; Friesland, M.; Khera, T.; Engelmann, M.; et al. Hepatitis E virus replication and interferon responses in human placental cells. Hepatol. Commun. 2018, 2, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.; Nguyen, H.T.; Torian, U.; Engle, R.E.; Faulk, K.; Dalton, H.R.; Bendall, R.P.; Keane, F.E.; Purcell, R.H.; Emerson, S.U. Cross-species infections of cultured cells by hepatitis E virus and discovery of an infectious virus-host recombinant. Proc. Natl. Acad. Sci. USA 2011, 108, 2438–2443. [Google Scholar] [CrossRef] [PubMed]
- Gouilly, J.; Chen, Q.; Siewiera, J.; Cartron, G.; Levy, C.; Dubois, M.; Al-Daccak, R.; Izopet, J.; Jabrane-Ferrat, N.; El Costa, H. Genotype specific pathogenicity of hepatitis E virus at the human maternal-fetal interface. Nat. Commun. 2018, 9, 4748. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Huang, F.; Xu, L.; Lin, Z.; de Vrij, F.M.S.; Ayo-Martin, A.C.; van der Kroeg, M.; Zhao, M.; Yin, Y.; Wang, W.; et al. Hepatitis E Virus Infects Neurons and Brains. J. Infect. Dis. 2017, 215, 1197–1206. [Google Scholar] [CrossRef] [PubMed]
- Fu, R.M.; Decker, C.C.; Dao Thi, V.L. Cell Culture Models for Hepatitis E Virus. Viruses 2019, 11, 608. [Google Scholar] [CrossRef] [PubMed]
- Meister, T.L.; Bruening, J.; Todt, D.; Steinmann, E. Cell culture systems for the study of hepatitis E virus. Antivir. Res. 2019, 163, 34–49. [Google Scholar] [CrossRef]
- Lemon, S.M.; Ott, J.J.; Van Damme, P.; Shouval, D. Type A viral hepatitis: A summary and update on the molecular virology, epidemiology, pathogenesis and prevention. J. Hepatol. 2018, 68, 167–184. [Google Scholar] [CrossRef]
- Okamoto, H. Culture systems for hepatitis E virus. J. Gastroenterol. 2013, 48, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Kalia, M.; Chandra, V.; Rahman, S.A.; Sehgal, D.; Jameel, S. Heparan Sulfate Proteoglycans Are Required for Cellular Binding of the Hepatitis E Virus ORF2 Capsid Protein and for Viral Infection. J. Virol. 2009, 83, 12714–12724. [Google Scholar] [CrossRef] [PubMed]
- Chu, H.; Chan, C.M.; Zhang, X.; Wang, Y.; Yuan, S.; Zhou, J.; Au-Yeung, R.K.; Sze, K.H.; Yang, D.; Shuai, H.; et al. Middle East respiratory syndrome coronavirus and bat coronavirus HKU9 both can utilize GRP78 for attachment onto host cells. J. Biol. Chem. 2018, 293, 11709–11726. [Google Scholar] [CrossRef] [PubMed]
- Nain, M.; Mukherjee, S.; Karmakar, S.P.; Paton, A.W.; Paton, J.C.; Abdin, M.Z.; Basu, A.; Kalia, M.; Vrati, S. GRP78 Is an Important Host Factor for Japanese Encephalitis Virus Entry and Replication in Mammalian Cells. J. Virol. 2017, 91, e02274-16. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Fradelizi, D.; Wilson, K.; Triantafilou, M. GRP78, a Coreceptor for Coxsackievirus A9, Interacts with Major Histocompatibility Complex Class I Molecules Which Mediate Virus Internalization. J. Virol. 2002, 76, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Honda, T.; Horie, M.; Daito, T.; Ikuta, K.; Tomonaga, K. Molecular Chaperone BiP Interacts with Borna Disease Virus Glycoprotein at the Cell Surface. J. Virol. 2009, 83, 12622–12625. [Google Scholar] [CrossRef]
- Yu, H.; Li, S.; Yang, C.; Wei, M.; Song, C.; Zheng, Z.; Gu, Y.; Du, H.; Zhang, J.; Xia, N. Homology model and potential virus-capsid binding site of a putative HEV receptor Grp78. J. Mol. Model. 2011, 17, 987–995. [Google Scholar] [CrossRef]
- Zhang, L.; Tian, Y.; Wen, Z.; Zhang, F.; Qi, Y.; Huang, W.; Zhang, H.; Wang, Y. Asialoglycoprotein receptor facilitates infection of PLC/PRF/5 cells by HEV through interaction with ORF2. J. Med. Virol. 2016, 88, 2186–2195. [Google Scholar] [CrossRef]
- Fongsaran, C.; Jirakanwisal, K.; Kuadkitkan, A.; Wikan, N.; Wintachai, P.; Thepparit, C.; Ubol, S.; Phaonakrop, N.; Roytrakul, S.; Smith, D.R. Involvement of ATP synthase beta subunit in chikungunya virus entry into insect cells. Arch. Virol. 2014, 159, 3353–3364. [Google Scholar] [CrossRef]
- Ahmed, Z.; Holla, P.; Ahmad, I.; Jameel, S. The ATP synthase subunit β (ATP5B) is an entry factor for the hepatitis E virus. bioRxiv 2016, 2016, 060434. [Google Scholar]
- Shiota, T.; Li, T.C.; Nishimura, Y.; Yoshizaki, S.; Sugiyama, R.; Shimojima, M.; Saijo, M.; Shimizu, H.; Suzuki, R.; Wakita, T.; et al. Integrin alpha3 is involved in non-enveloped hepatitis E virus infection. Virology 2019, 536, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Kapur, N.; Thakral, D.; Durgapal, H.; Panda, S.K. Hepatitis E virus enters liver cells through receptor-dependent clathrin-mediated endocytosis. J. Viral Hepat. 2012, 19, 436–448. [Google Scholar] [CrossRef] [PubMed]
- Holla, P.; Ahmad, I.; Ahmed, Z.; Jameel, S. Hepatitis E Virus Enters Liver Cells Through a Dynamin-2, Clathrin and Membrane Cholesterol-Dependent Pathway. Traffic 2015, 16, 398–416. [Google Scholar] [CrossRef] [PubMed]
- Bubeck, D.; Filman, D.J.; Cheng, N.; Steven, A.C.; Hogle, J.M.; Belnap, D.M. The structure of the poliovirus 135S cell entry intermediate at 10-angstrom resolution reveals the location of an externalized polypeptide that binds to membranes. J. Virol. 2005, 79, 7745–7755. [Google Scholar] [CrossRef] [PubMed]
- Jemielity, S.; Wang, J.J.; Chan, Y.K.; Ahmed, A.A.; Li, W.; Monahan, S.; Bu, X.; Farzan, M.; Freeman, G.J.; Umetsu, D.T. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 2013, 9, e1003232. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Maury, W.; Lemon, S.M. TIM1 (HAVCR1): an Essential "Receptor" or an "Accessory Attachment Factor" for Hepatitis A Virus? J. Virol. 2019, 93. [Google Scholar] [CrossRef]
- Pischke, S.; Hartl, J.; Pas, S.D.; Lohse, A.W.; Jacobs, B.C.; Van der Eijk, A.A. Hepatitis E virus: Infection beyond the liver? J. Hepatol. 2017, 66, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Rivera-Serrano, E.E.; Gonzalez-Lopez, O.; Das, A.; Lemon, S.M. Cellular entry and uncoating of naked and quasi-enveloped human hepatoviruses. Elife 2019, 8, e43983. [Google Scholar] [CrossRef]
- Kolter, T.; Sandhoff, K. Lysosomal degradation of membrane lipids. FEBS Lett. 2010, 584, 1700–1712. [Google Scholar] [CrossRef]
- Dubland, J.A.; Francis, G.A. Lysosomal acid lipase: At the crossroads of normal and atherogenic cholesterol metabolism. Front. Cell Dev. Biol. 2015, 3, 3. [Google Scholar] [CrossRef]
- Moller-Tank, S.; Maury, W. Ebola virus entry: A curious and complex series of events. PLoS Pathog. 2015, 11, e1004731. [Google Scholar] [CrossRef] [PubMed]
- Coyne, C.B.; Bergelson, J.M. Virus-induced Abl and Fyn kinase signals permit coxsackievirus entry through epithelial tight junctions. Cell 2006, 124, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, S.; Korkaya, H.; Zafrullah, M.; Jameel, S.; Lal, S.K. The phosphorylated form of the ORF3 protein of hepatitis E virus interacts with its non-glycosylated form of the major capsid protein, ORF2. J. Biol. Chem. 2002, 277, 22759–22767. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Heller, B.; Capuccino, J.M.; Song, B.; Nimgaonkar, I.; Hrebikova, G.; Contreras, J.E.; Ploss, A. Hepatitis E virus ORF3 is a functional ion channel required for release of infectious particles. Proc. Natl. Acad. Sci. USA 2017, 114, 1147–1152. [Google Scholar] [CrossRef] [PubMed]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, X.; Feng, Z. Hepatitis E Virus Entry. Viruses 2019, 11, 883. https://doi.org/10.3390/v11100883
Yin X, Feng Z. Hepatitis E Virus Entry. Viruses. 2019; 11(10):883. https://doi.org/10.3390/v11100883
Chicago/Turabian StyleYin, Xin, and Zongdi Feng. 2019. "Hepatitis E Virus Entry" Viruses 11, no. 10: 883. https://doi.org/10.3390/v11100883
APA StyleYin, X., & Feng, Z. (2019). Hepatitis E Virus Entry. Viruses, 11(10), 883. https://doi.org/10.3390/v11100883