Rates of Molecular Evolution in a Marine Synechococcus Phage Lineage


Abstract
1. Introduction
2. Materials and Methods
3. Results
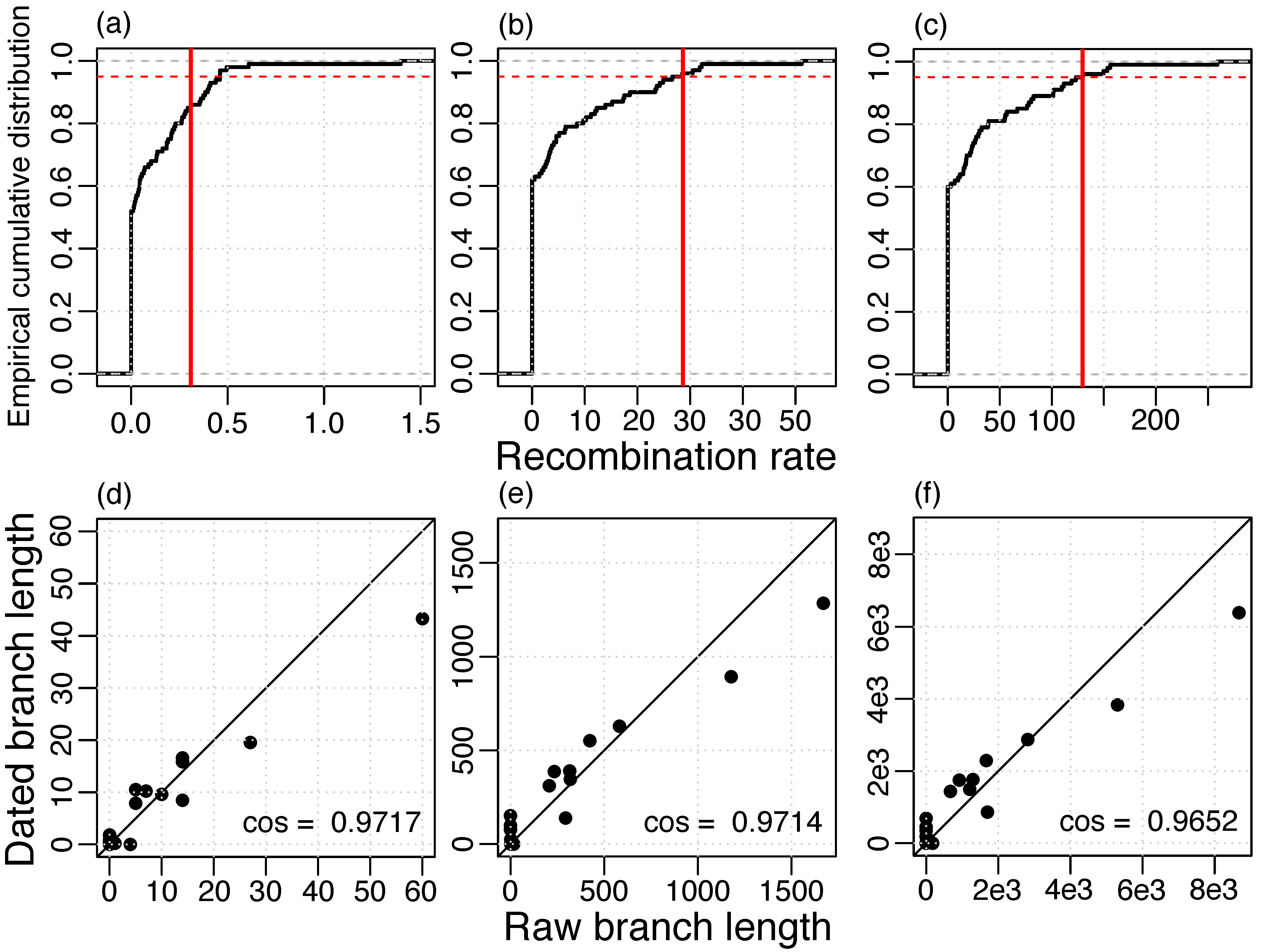
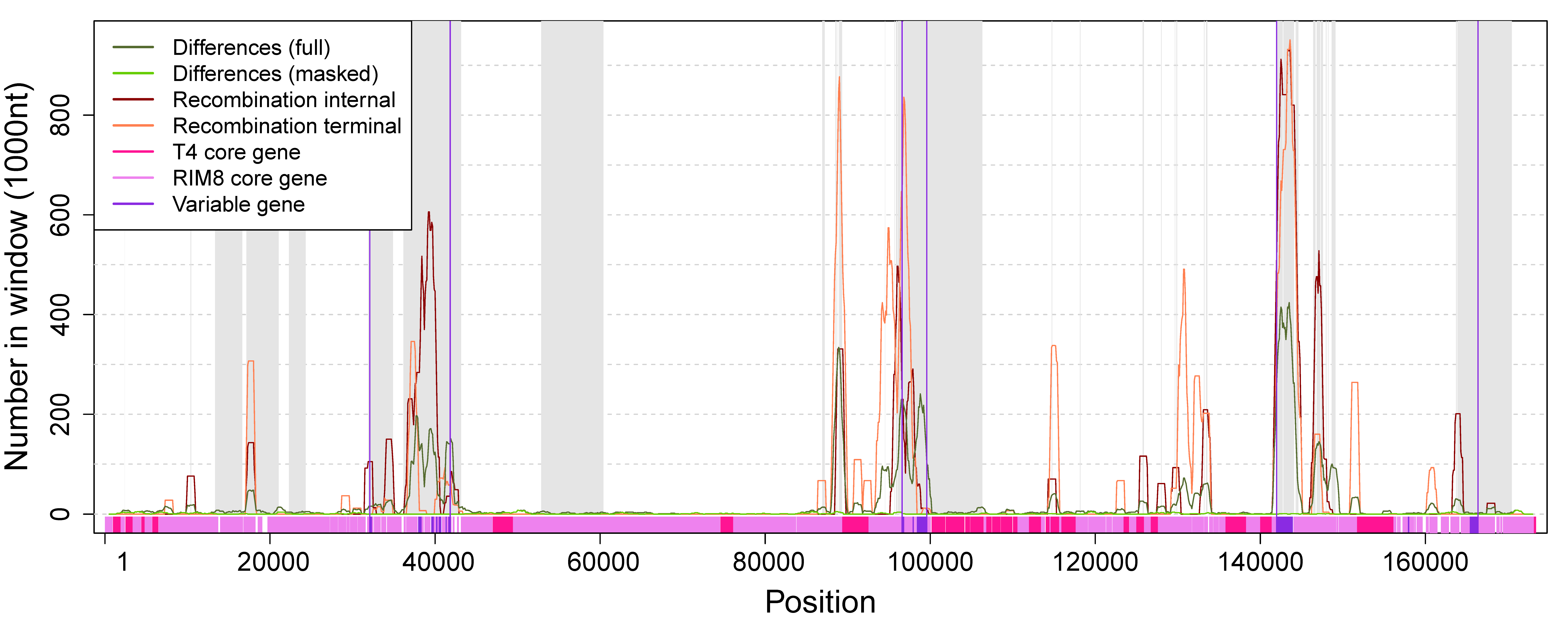
3.1. Recombination Detection
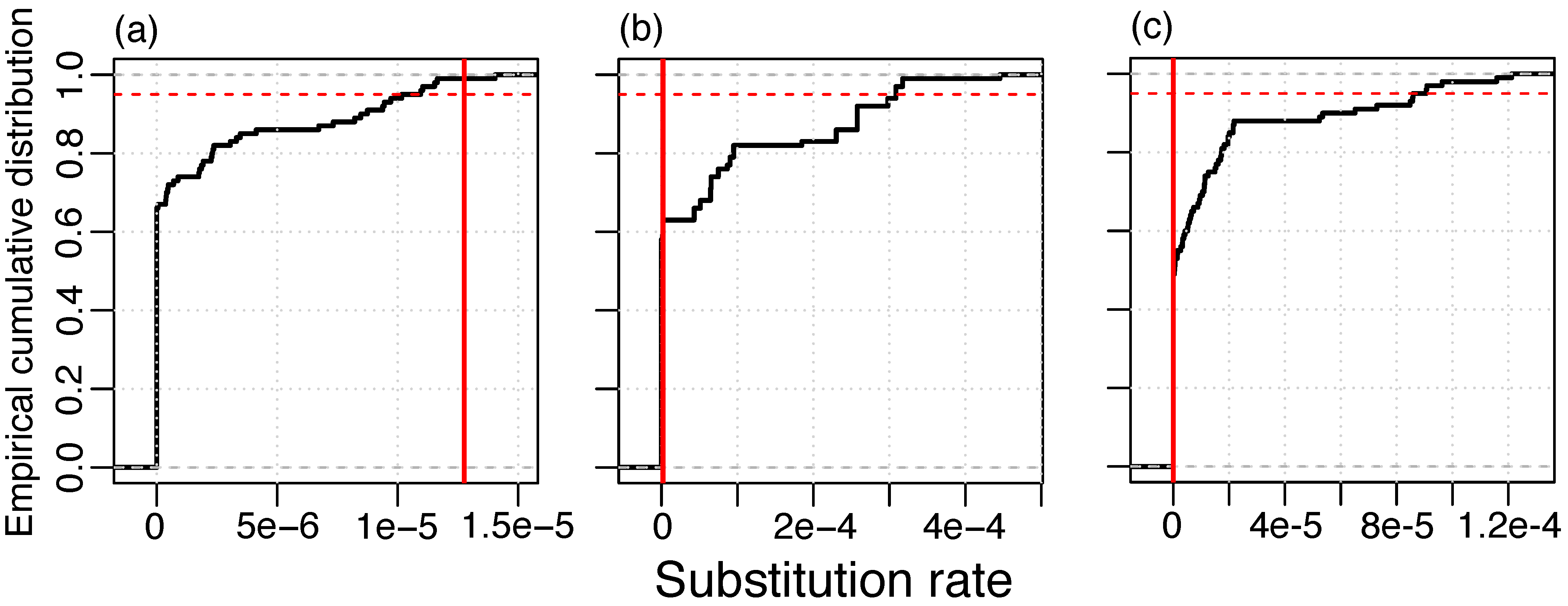
3.2. Temporal Signal and Substitution Rate Estimation
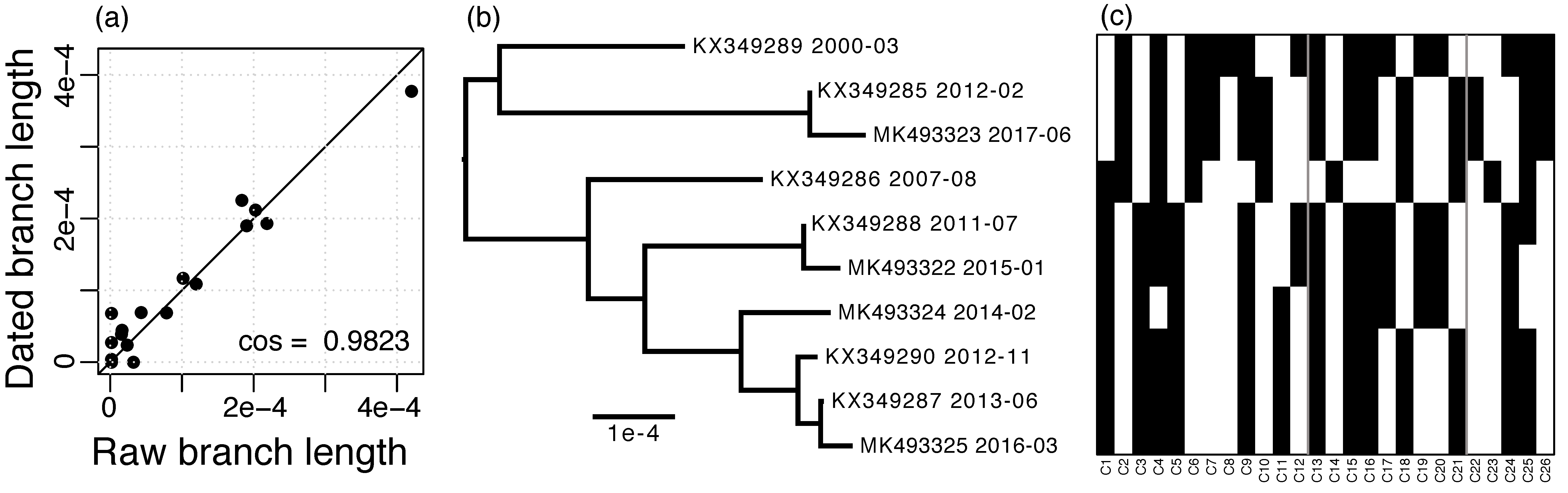
3.3. RIM8 Evolutionary Rates
3.4. Variable Gene Families
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mühling, M.; Fuller, N.J.; Millard, A.; Somerfield, P.J.; Marie, D.; Wilson, W.H.; Scanlan, D.J.; Post, A.F.; Joint, I.; Mann, N.H. Genetic diversity of marine Synechococcus and co-occurring cyanophage communities: Evidence for viral control of phytoplankton. Environ. Microbiol. 2005, 7, 499–508. [Google Scholar] [CrossRef]
- Bouvier, T.; Del Giorgio, P.A. Key role of selective viral-induced mortality in determining marine bacterial community composition. Environ. Microbiol. 2007, 9, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Suttle, C.A. Marine viruses–major players in the global ecosystem. Nat. Rev. Microbiol. 2007, 5, 801–812. [Google Scholar] [CrossRef]
- Breitbart, M.; Bonnain, C.; Malki, K.; Sawaya, N.A. Phage puppet masters of the marine microbial realm. Nat. Microbiol. 2018, 3, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Warwick-Dugdale, J.; Buchholz, H.H.; Allen, M.J.; Temperton, B. Host-hijacking and planktonic piracy: How phages command the microbial high seas. Virol. J. 2019, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Coleman, M.L.; Sullivan, M.B.; Martiny, A.C.; Steglich, C.; Barry, K.; DeLong, E.F.; Chisholm, S.W. Genomic islands and the ecology and evolution of Prochlorococcus. Science 2006, 311, 1768–1770. [Google Scholar] [CrossRef] [PubMed]
- Lindell, D.; Jaffe, J.D.; Johnson, Z.I.; Church, G.M.; Chisholm, S.W. Photosynthesis genes in marine viruses yield proteins during host infection. Nature 2005, 438, 86–89. [Google Scholar] [CrossRef] [PubMed]
- Sieradzki, E.T.; Ignacio-Espinoza, J.C.; Needham, D.M.; Fichot, E.B.; Fuhrman, J.A. Dynamic marine viral infections and major contribution to photosynthetic processes shown by spatiotemporal picoplankton metatranscriptomes. Nat. Commun. 2019, 10, 1169. [Google Scholar] [CrossRef]
- Puxty, R.J.; Millard, A.D.; Evans, D.J.; Scanlan, D.J. Shedding new light on viral photosynthesis. Photosynth. Res. 2015, 126, 71–97. [Google Scholar] [CrossRef]
- Schwartz, D.A.; Lindell, D. Genetic hurdles limit the arms race between Prochlorococcus and the T7-like podoviruses infecting them. ISME J. 2017, 11, 1836–1851. [Google Scholar] [CrossRef]
- Enav, H.; Kirzner, S.; Lindell, D.; Mandel-Gutfreund, Y.; Béjà, O. Adaptation to sub-optimal hosts is a driver of viral diversification in the ocean. Nat. Commun. 2018, 9, 4698. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.W. A constant rate of spontaneous mutation in DNA-based microbes. Proc. Natl. Acad. Sci. USA 1991, 88, 7160–7164. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R.; Nebot, M.R.; Chirico, N.; Mansky, L.M.; Belshaw, R. Viral mutation rates. J. Virol. 2010, 84, 9733–9748. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Pybus, O.G.; Rambaut, A.; Forsberg, R.; Rodrigo, A.G. Measurably evolving populations. Trends Ecol. Evol. 2003, 18, 481–488. [Google Scholar] [CrossRef]
- Rieux, A.; Balloux, F. Inferences from tip-calibrated phylogenies: A review and a practical guide. Mol. Ecol. 2016, 25, 1911–1924. [Google Scholar] [CrossRef] [PubMed]
- Duchêne, S.; Holt, K.E.; Weill, F.-X.; Le Hello, S.; Hawkey, J.; Edwards, D.J.; Fourment, M.; Holmes, E.C. Genome-scale rates of evolutionary change in bacteria. Microb. Genomics 2016, 2, e000094. [Google Scholar] [CrossRef] [PubMed]
- Biek, R.; Pybus, O.G.; Lloyd-Smith, J.O.; Didelot, X. Measurably evolving pathogens in the genomic era. Trends Ecol. Evol. 2015, 30, 306–313. [Google Scholar] [CrossRef]
- Kupczok, A.; Neve, H.; Huang, K.D.; Hoeppner, M.P.; Heller, K.J.; Franz, C.M.A.P.; Dagan, T. Rates of mutation and recombination in Siphoviridae phage genome evolution over three decades. Mol. Biol. Evol. 2018, 35, 1147–1159. [Google Scholar] [CrossRef]
- Díaz-Muñoz, S.L. Viral coinfection is shaped by host ecology and virus–virus interactions across diverse microbial taxa and environments. Virus Evol. 2017, 3, vex011. [Google Scholar] [CrossRef]
- Roux, S.; Hawley, A.K.; Torres Beltran, M.; Scofield, M.; Schwientek, P.; Stepanauskas, R.; Woyke, T.; Hallam, S.J.; Sullivan, M.B. Ecology and evolution of viruses infecting uncultivated SUP05 bacteria as revealed by single-cell- and meta-genomics. eLife 2014, 3, e03125. [Google Scholar] [CrossRef]
- Millard, A.D.; Zwirglmaier, K.; Downey, M.J.; Mann, N.H.; Scanlan, D.J. Comparative genomics of marine cyanomyoviruses reveals the widespread occurrence of Synechococcus host genes localized to a hyperplastic region: Implications for mechanisms of cyanophage evolution. Environ. Microbiol. 2009, 11, 2370–2387. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B.; Huang, K.H.; Ignacio-Espinoza, J.C.; Berlin, A.M.; Kelly, L.; Weigele, P.R.; DeFrancesco, A.S.; Kern, S.E.; Thompson, L.R.; Young, S.; et al. Genomic analysis of oceanic cyanobacterial myoviruses compared with T4-like myoviruses from diverse hosts and environments. Environ. Microbiol. 2010, 12, 3035–3056. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Morrical, S.W. Assembly and dynamics of the bacteriophage T4 homologous recombination machinery. Virol. J. 2010, 7, 357. [Google Scholar] [CrossRef] [PubMed]
- Marston, M.F.; Amrich, C.G. Recombination and microdiversity in coastal marine cyanophages. Environ. Microbiol. 2009, 11, 2893–2903. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.B.; Lindell, D.; Lee, J.A.; Thompson, L.R.; Bielawski, J.P.; Chisholm, S.W. Prevalence and Evolution of Core Photosystem II Genes in Marine Cyanobacterial Viruses and Their Hosts. PLoS Biol. 2006, 4, e234. [Google Scholar] [CrossRef] [PubMed]
- Comeau, A.M.; Bertrand, C.; Letarov, A.; Tétart, F.; Krisch, H.M. Modular architecture of the T4 phage superfamily: A conserved core genome and a plastic periphery. Virology 2007, 362, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Cordero, O.X.; Polz, M.F. Explaining microbial genomic diversity in light of evolutionary ecology. Nat. Rev. Microbiol. 2014, 12, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.C.; Solonenko, S.A.; Ignacio-Espinoza, J.C.; LaButti, K.; Copeland, A.; Sudek, S.; Maitland, A.; Chittick, L.; dos Santos, F.; Weitz, J.S.; et al. Genomic differentiation among wild cyanophages despite widespread horizontal gene transfer. BMC Genomics 2016, 17, 930. [Google Scholar] [CrossRef] [PubMed]
- Marston, M.F.; Martiny, J.B.H. Genomic diversification of marine cyanophages into stable ecotypes: Cyanophage diversification into ecotypes. Environ. Microbiol. 2016, 18, 4240–4253. [Google Scholar] [CrossRef]
- Ignacio-Espinoza, J.C.; Sullivan, M.B. Phylogenomics of T4 cyanophages: Lateral gene transfer in the ‘core’ and origins of host genes. Environ. Microbiol. 2012, 14, 2113–2126. [Google Scholar] [CrossRef]
- Cordero, O.X. Endemic cyanophages and the puzzle of phage-bacteria coevolution. Environ. Microbiol. 2017, 19, 420–422. [Google Scholar] [CrossRef] [PubMed]
- Darling, A.E.; Mau, B.; Perna, N.T. progressiveMauve: Multiple genome alignment with gene gain, loss and rearrangement. PLoS ONE 2010, 5, e11147. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast model selection for accurate phylogenetic estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Wilson, D.J. ClonalFrameML: Efficient inference of recombination in whole bacterial genomes. PLoS Comput. Biol. 2015, 11, e1004041. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef] [PubMed]
- To, T.-H.; Jung, M.; Lycett, S.; Gascuel, O. Fast Dating Using Least-Squares Criteria and Algorithms. Syst. Biol. 2016, 65, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-likelihood phylodynamic analysis. Virus Evol. 2018, 4, vex042. [Google Scholar] [CrossRef]
- Hope, A.B. Electron transfers amongst cytochrome f, plastocyanin and photosystem I: Kinetics and mechanisms. Biochim. Biophys. Acta BBA Bioenerget. 2000, 1456, 5–26. [Google Scholar] [CrossRef]
- Crummett, L.T.; Puxty, R.J.; Weihe, C.; Marston, M.F.; Martiny, J.B.H. The genomic content and context of auxiliary metabolic genes in marine cyanomyoviruses. Virology 2016, 499, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Nakayashiki, T.; Mori, H. Genome-Wide Screening with Hydroxyurea Reveals a Link between Nonessential Ribosomal Proteins and Reactive Oxygen Species Production. J. Bacteriol. 2013, 195, 1226–1235. [Google Scholar] [CrossRef] [PubMed]
- Soule, T.; Gao, Q.; Stout, V.; Garcia-Pichel, F. The Global Response of Nostoc punctiforme ATCC 29133 to UVA Stress, Assessed in a Temporal DNA Microarray Study. Photochem. Photobiol. 2013, 89, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Sung, W.; Ackerman, M.S.; Miller, S.F.; Doak, T.G.; Lynch, M. Drift-barrier hypothesis and mutation-rate evolution. Proc. Natl. Acad. Sci. USA 2012, 109, 18488–18492. [Google Scholar] [CrossRef] [PubMed]
- Sanjuán, R. From Molecular Genetics to Phylodynamics: Evolutionary Relevance of Mutation Rates Across Viruses. PLoS Pathog. 2012, 8, e1002685. [Google Scholar] [CrossRef] [PubMed]
- Ohta, T.; Kimura, M. On the constancy of the evolutionary rate of cistrons. J. Mol. Evol. 1971, 1, 18–25. [Google Scholar] [CrossRef]
- Duchêne, S.; Holmes, E.C. Estimating evolutionary rates in giant viruses using ancient genomes. Virus Evol. 2018, 4. [Google Scholar] [CrossRef]
- Marston, M.F.; Pierciey, F.J.; Shepard, A.; Gearin, G.; Qi, J.; Yandava, C.; Schuster, S.C.; Henn, M.R.; Martiny, J.B.H. Rapid diversification of coevolving marine Synechococcus and a virus. Proc. Natl. Acad. Sci. USA 2012, 109, 4544–4549. [Google Scholar] [CrossRef]
- Croucher, N.J.; Harris, S.R.; Fraser, C.; Quail, M.A.; Burton, J.; van der Linden, M.; McGee, L.; von Gottberg, A.; Song, J.H.; Ko, K.S.; et al. Rapid pneumococcal evolution in response to clinical interventions. Science 2011, 331, 430–434. [Google Scholar] [CrossRef]
- Dettman, J.R.; Rodrigue, N.; Kassen, R. Genome-Wide Patterns of Recombination in the Opportunistic Human Pathogen Pseudomonas aeruginosa. Genome Biol. Evol. 2015, 7, 18–34. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Méric, G.; Falush, D.; Darling, A.E. Impact of homologous and non-homologous recombination in the genomic evolution of Escherichia coli. BMC Genomics 2012, 13, 256. [Google Scholar] [CrossRef] [PubMed]
- Didelot, X.; Bowden, R.; Street, T.; Golubchik, T.; Spencer, C.; McVean, G.; Sangal, V.; Anjum, M.F.; Achtman, M.; Falush, D.; et al. Recombination and Population Structure in Salmonella enterica. PLoS Genet. 2011, 7, e1002191. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Didelot, X. Inference of the properties of the recombination process from whole bacterial genomes. Genetics 2013, 196, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Mizuno, C.M.; Ghai, R.; Rodriguez-Valera, F. Evidence for metaviromic islands in marine phages. Front. Microbiol. 2014, 5, 27. [Google Scholar] [CrossRef] [PubMed]
- Mann, N.H. Phages of the marine cyanobacterial picophytoplankton. FEMS Microbiol. Rev. 2003, 27, 17–34. [Google Scholar] [CrossRef]
- Fridman, S.; Flores-Uribe, J.; Larom, S.; Alalouf, O.; Liran, O.; Yacoby, I.; Salama, F.; Bailleul, B.; Rappaport, F.; Ziv, T.; et al. A myovirus encoding both photosystem I and II proteins enhances cyclic electron flow in infected Prochlorococcus cells. Nat. Microbiol. 2017, 2, 1350–1357. [Google Scholar] [CrossRef]
- Puxty, R.J.; Evans, D.J.; Millard, A.D.; Scanlan, D.J. Energy limitation of cyanophage development: Implications for marine carbon cycling. ISME J. 2018, 12, 1273–1286. [Google Scholar] [CrossRef]
- Kelly, L.; Ding, H.; Huang, K.H.; Osburne, M.S.; Chisholm, S.W. Genetic diversity in cultured and wild marine cyanomyoviruses reveals phosphorus stress as a strong selective agent. ISME J. 2013, 7, 1827–1841. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | Number of Genomes | Time Span (Years) | Number of Timepoints | Mean Length (nt) | p-Value Phi Test | r/m |
|---|---|---|---|---|---|---|
| RIM2 | 59 | 8 | 9 | 175,301 | 6.01 × 10−34 | 2.156 |
| RIM2_A | 47 | 8 | 8 | 175,310 | 3.94 × 10−27 | 1.574 |
| RIM2_B | 10 | 1 | 4 | 175,309 | 5.24 × 10−9 | 3.385 |
| RIM8 | 10 | 17 | 10 | 170,485 | 0.0151 | 7.833 |
| RIM12 | 21 | 13 | 9 | 174,726 | 5.98 × 10−10 | 8.421 |
| RIM12_A | 10 | 13 | 7 | 174,271 | 2.07 × 10−12 | 7.053 |
| RIM12_C | 7 | 1 | 3 | 175,605 | 0.013 | 15.37 |
| RIM14 | 9 | 2 | 6 | 179,756 | 9.79 × 10−8 | 2.214 |
| RIM44 | 8 | 7 | 3 | 195,353 | 0.059 | 0.2884 |
| Name | Group | r/m | Reference |
|---|---|---|---|
| Staphylococcus aureus | Bacteria | 0.283 | [36] |
| Pseudomonas aeruginosa | Bacteria | 0.853 | [51] |
| Escherichia coli | Bacteria | 1.024 | [52] |
| Salmonella enterica | Bacteria | 1.14 | [53] |
| Bacillus cereus | Bacteria | 3.4 | [54] |
| Streptococcus pneumonia | Bacteria | 7.2 | [50] |
| Cyanophage RIM8 | dsDNA phage | 7.8 | This study |
| 936 group of phages | dsDNA phage | 23.5 | [18] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kupczok, A.; Dagan, T. Rates of Molecular Evolution in a Marine Synechococcus Phage Lineage. Viruses 2019, 11, 720. https://doi.org/10.3390/v11080720
Kupczok A, Dagan T. Rates of Molecular Evolution in a Marine Synechococcus Phage Lineage. Viruses. 2019; 11(8):720. https://doi.org/10.3390/v11080720
Chicago/Turabian StyleKupczok, Anne, and Tal Dagan. 2019. "Rates of Molecular Evolution in a Marine Synechococcus Phage Lineage" Viruses 11, no. 8: 720. https://doi.org/10.3390/v11080720
APA StyleKupczok, A., & Dagan, T. (2019). Rates of Molecular Evolution in a Marine Synechococcus Phage Lineage. Viruses, 11(8), 720. https://doi.org/10.3390/v11080720